N-terminal oligomerization drives HDAC4 nuclear condensation and neurodevelopmental dysfunction in Drosophila
Hannah R. Hawley, Andrew J. Sutherland-Smith, Matthew S. Savoian, Helen L. Fitzsimons

TL;DR
This study shows that HDAC4 protein forms nuclear condensates in fruit flies, which disrupts brain development and suggests targeting these structures could help treat related disorders.
Contribution
The study reveals that HDAC4 nuclear condensation is driven by self-oligomerization and worsened by MEF2, offering new therapeutic strategies for neurodevelopmental diseases.
Findings
HDAC4 nuclear condensation correlates with neurodevelopmental defects in Drosophila mushroom body and eye development.
Blocking HDAC4 oligomerization reduces condensation and severity of neurodevelopmental phenotypes.
MEF2 stabilizes HDAC4 condensates and increases phenotypic severity.
Abstract
Histone deacetylase four (HDAC4) undergoes dynamic nucleocytoplasmic shuttling, a process critical for regulating its activity. However, aberrant nuclear accumulation of HDAC4 is associated with both neurodevelopmental and neurodegenerative disease, and in our Drosophila model, impairs normal neuronal development. Upon nuclear accumulation, HDAC4 forms biomolecular condensates, previously termed aggregates, that correlate with the severity of defects in development of the Drosophila mushroom body and adult eye. Here we determined that nuclear condensation of HDAC4 is dependent on self-oligomerization, and that impairing oligomerization reduces condensation and the severity of neurodevelopmental phenotypes in Drosophila. HDAC4 condensates are highly dynamic and are stabilized by the presence of MEF2, which promotes their formation, ultimately exacerbating phenotypic severity. These data…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
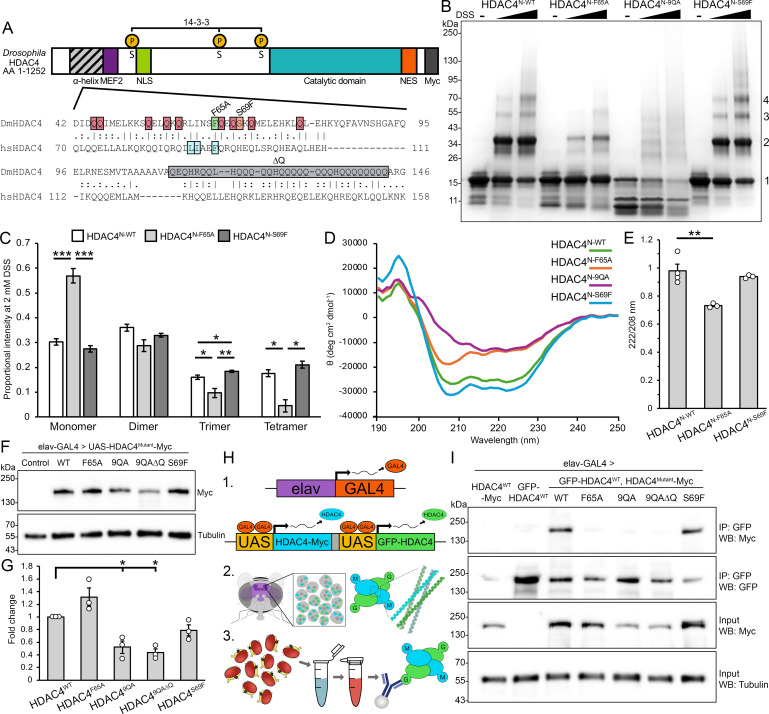 Figure 1
Figure 1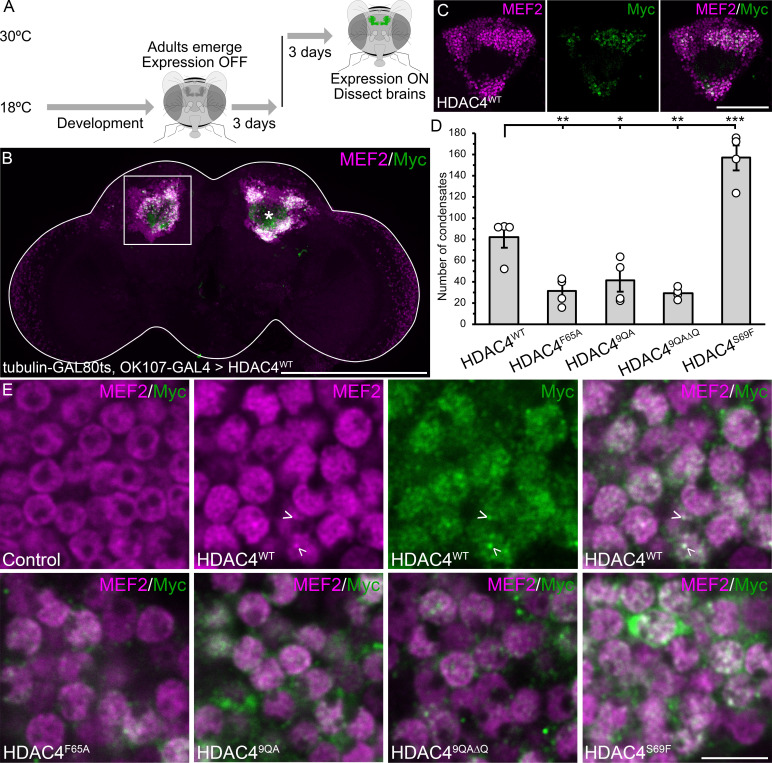 Figure 2
Figure 2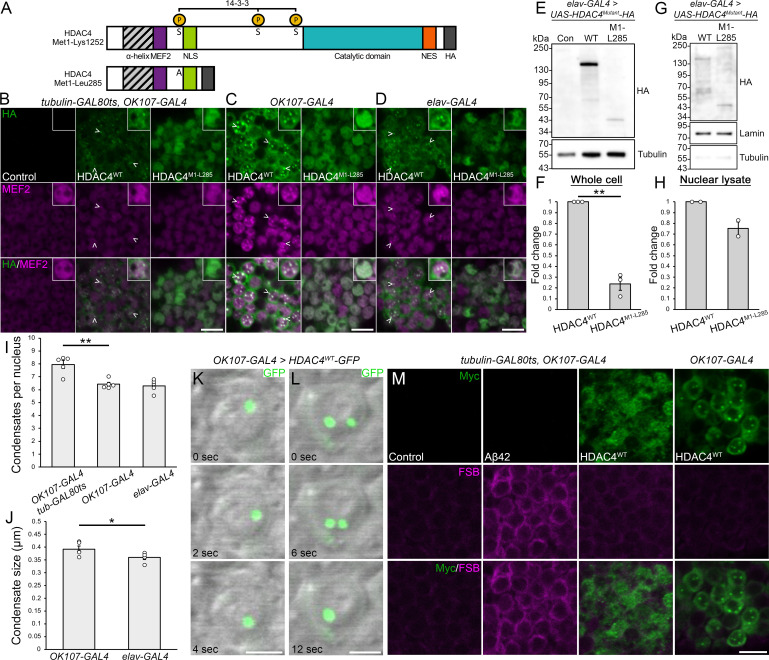 Figure 3
Figure 3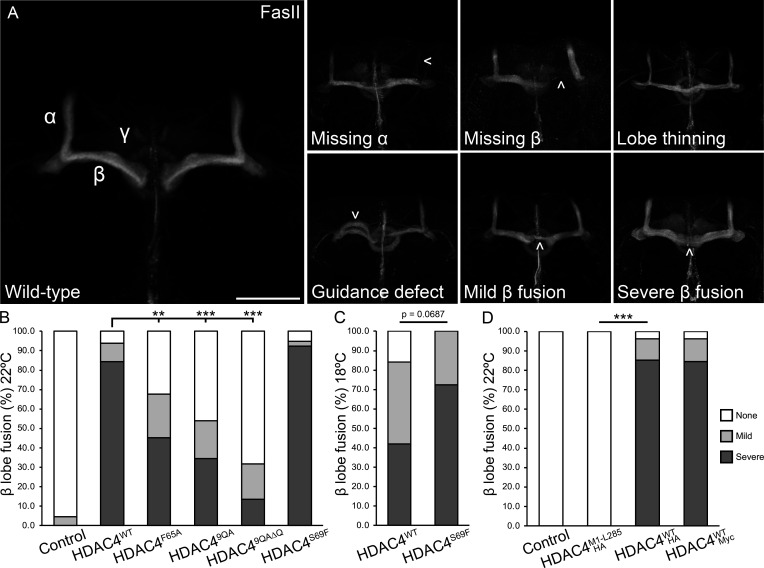 Figure 4
Figure 4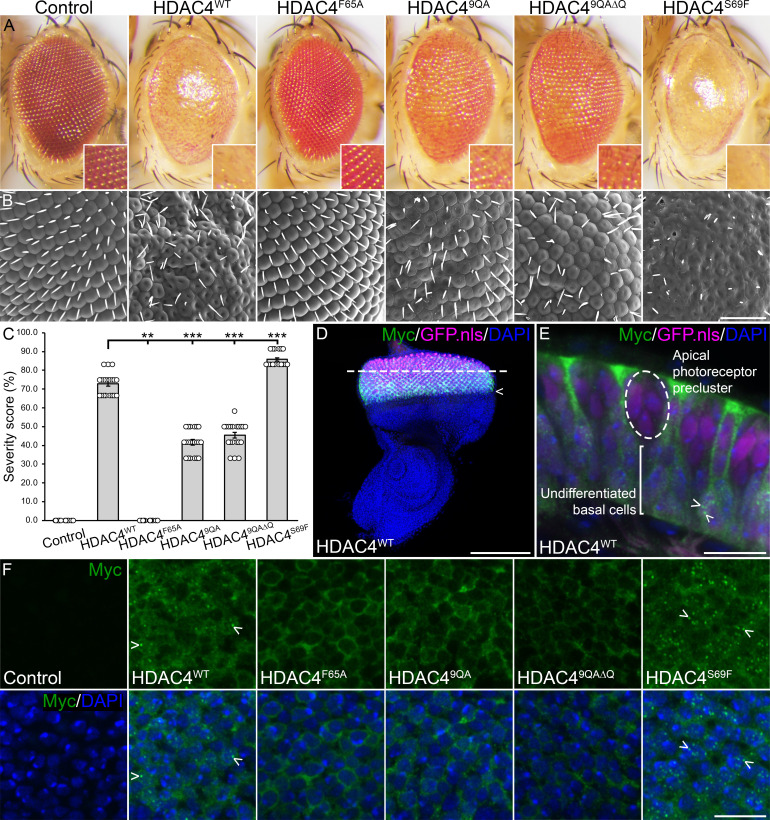 Figure 5
Figure 5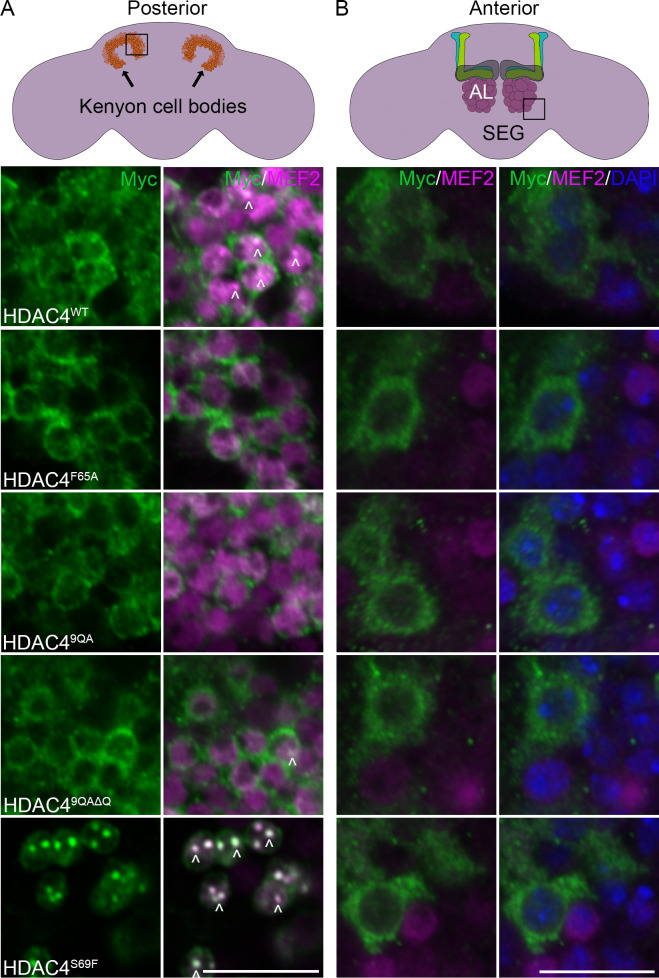 Figure 6
Figure 6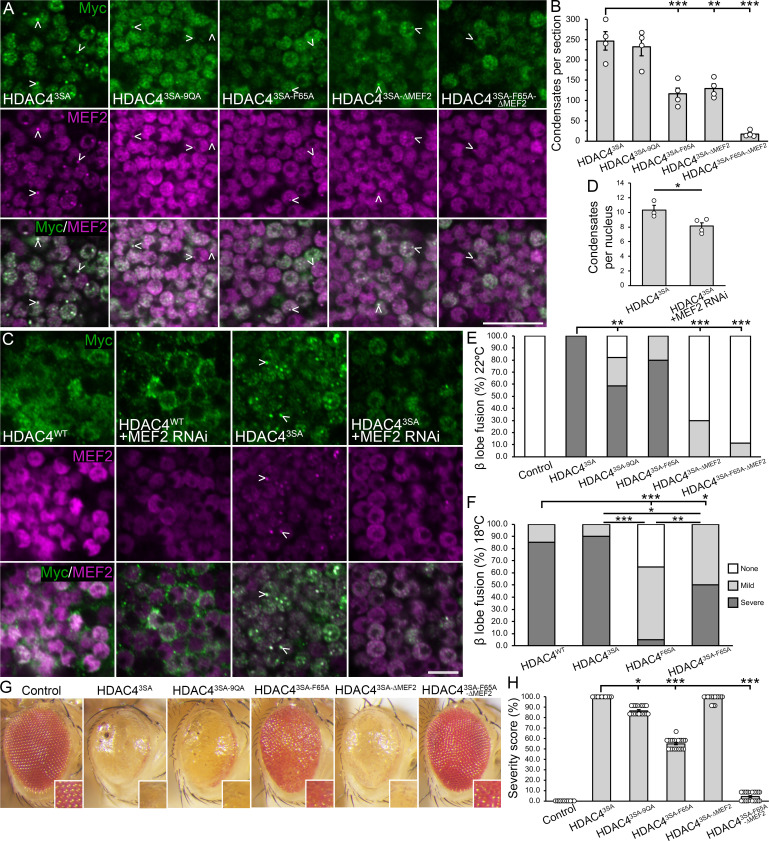 Figure 7
Figure 7|
22°C |
18°C | |||||||
|---|---|---|---|---|---|---|---|---|
|
(% of brains) |
control |
HDAC4WT |
HDAC4F65A |
HDAC49QA |
HDAC49QA∆Q |
HDAC4S69F |
HDAC4WT |
HDAC4S69F |
|
α lobe thinning |
0 |
6 |
0 |
8 |
0 |
26 |
5 |
10 |
|
α lobe missing |
0 |
0 |
0 |
4 |
0 |
5 |
0 |
0 |
|
β lobe thinning |
0 |
3 |
0 |
8 |
0 |
13 |
11 |
3 |
|
β lobe missing |
0 |
0 |
0 |
0 |
0 |
3 |
5 |
0 |
|
β lobe fusion |
5 |
94 |
68 |
54 |
32 |
95 |
84 |
100 |
|
mild |
5 |
9 |
23 |
19 |
18 |
3 |
42 |
28 |
|
severe |
0 |
84 |
45 |
35 |
14 |
92 |
42 |
72 |
|
guidance defect |
0 |
3 |
0 |
0 |
4 |
0 |
5 |
0 |
|
no defects |
95 |
3 |
32 |
42 |
64 |
3 |
11 |
0 |
|
|
44 |
32 |
31 |
26 |
22 |
39 |
19 |
29 |
|
22°C | ||||
|---|---|---|---|---|
|
(% of brains) |
control |
HDAC4M1-L285-HA |
HDAC4WT-HA |
HDAC4WT-Myc |
|
α lobe thinning |
4 |
0 |
59 |
35 |
|
α lobe missing |
0 |
0 |
0 |
4 |
|
β lobe thinning |
0 |
0 |
33 |
0 |
|
β lobe missing |
0 |
0 |
0 |
4 |
|
β lobe fusion |
0 |
0 |
97 |
96 |
|
mild |
0 |
0 |
12 |
11 |
|
severe |
0 |
0 |
85 |
85 |
|
guidance defect |
0 |
0 |
4 |
0 |
|
no defects |
100 |
100 |
0 |
4 |
|
|
23 |
22 |
27 |
26 |
|
22°C |
18°C | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
(% of brains) |
control |
HDAC4 3SA |
HDAC4 3SA-9QA |
HDAC4 3SA-F65A |
HDAC4 3SA-∆MEF2 |
HDAC4 3SA-F65A-∆MEF2 |
control |
HDAC4 WT |
HDAC4 3SA |
HDAC4 F65A |
HDAC4 3SA-F65A |
|
α lobe thinning |
0 |
50 |
27 |
35 |
5 |
0 |
0 |
10 |
40 |
0 |
10 |
|
α lobe missing |
0 |
6 |
24 |
0 |
0 |
0.0 |
5 |
0 |
5 |
0 |
0 |
|
β lobe thinning |
0 |
13 |
9 |
5 |
0 |
6 |
0 |
0 |
10 |
0 |
15 |
|
β lobe missing |
0 |
13 |
0 |
5 |
5 |
11 |
0 |
0 |
5 |
10 |
5 |
|
β lobe fusion |
0 |
100 |
82 |
100 |
30 |
11 |
20 |
100 |
100 |
65 |
100 |
|
mild |
0 |
0 |
24 |
20 |
30 |
11 |
20 |
15 |
10 |
60 |
50 |
|
severe |
0 |
100 |
59 |
80 |
0 |
0 |
0 |
85 |
90 |
5 |
50 |
|
guidance defect |
0 |
50 |
32 |
45 |
10 |
11 |
5 |
0 |
50 |
15 |
40 |
|
no defects |
100 |
0 |
6 |
0 |
60 |
78 |
75 |
0 |
0 |
25 |
0 |
|
|
20 |
16 |
34 |
20 |
20 |
18 |
20 |
20 |
20 |
20 |
20 |
- —Marsden Fundhttp://dx.doi.org/10.13039/501100009193
- —Palmerston North Medical Research Foundationhttp://dx.doi.org/10.13039/100012500
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHistone Deacetylase Inhibitors Research · Signaling Pathways in Disease · Genetics and Neurodevelopmental Disorders
Introduction
Histone deacetylase four (HDAC4) is an important regulator of neuronal development and memory formation in species across the animal kingdom including C. elegans, Drosophila, rodents and humans [1–7]. In mammals, HDAC4 is expressed widely in the brain [8–10]. As a member of the Class IIa family of histone deacetylases, it is characterized by a conserved deacetylase domain and an extended N-terminal region that interacts with regulatory proteins including transcription factors [11–14]. Although it contains a deacetylase domain, vertebrate HDAC4 harbours little intrinsic deacetylase activity [15], instead facilitating deacetylation via association with HDAC3 and the NCoR/SMRT repressor complex [16–18].
HDAC4 shuttles between the nucleus and cytoplasm in a phosphorylation-dependent manner. Nuclear export is mediated by 14-3-3 binding to phosphorylated serines (Ser246, Ser467 and Ser632 in human HDAC4) [19–22], while nuclear import is mediated through interaction with the transcription factor MEF2 [14,23]. This dynamic regulation is governed by synaptic activity and differs between neuronal subtypes [3,5,9,24–27].
Aberrant nuclear accumulation of HDAC4 has been associated with several neurodegenerative and neurodevelopmental disorders. Increased nuclear concentration of HDAC4 has been observed in the brains of individuals with Alzheimer’s disease (AD) [28,29] and ataxia telangiectasia [30], as well as in mouse models of AD [28,31,32], CDKL5 disorder [7], 2q37 deletion syndrome [3] and Parkinson’s disease [33,34]. Mutations within the 14-3-3 binding site of HDAC4 have been identified in seven unrelated individuals with intellectual disability and developmental delay, and these mutations reduce the affinity of HDAC4 for 14-3-3 in cultured cells [6]. Consequently, it is hypothesized that disrupted nucleocytoplasmic shuttling of HDAC4 underlies neuronal dysfunction in these individuals.
HDAC4 staining in neurons displays a punctate, granular pattern [9,35], which is thought to involve self-oligomerization mediated by the N-terminal helix [36,37]. When HDAC4 accumulates in neuronal nuclei it forms larger punctate foci [32,34,38], which have been variously described as speckles or aggregates. Recent evidence indicates that HDAC4 foci are more accurately described as biomolecular condensates (termed condensates herein) [39], which are dynamic subnuclear compartments that regulate molecular interactions [40]. Disruption of these condensates, as observed in neuronal disease, is therefore likely to contribute to neuronal dysfunction. Despite numerous observations of nuclear HDAC4 accumulation in neuronal disease, the molecular mechanisms driving condensate formation and their contribution to disease onset and/or progression remain unexplored, highlighting a critical gap in understanding.
Drosophila HDAC4 shares key regulatory motifs with human HDAC4, including nuclear localization/export signals, the MEF2 binding region, catalytic site, ankyrin repeat binding region and conserved phosphorylation sites [4,35]. HDAC4 is highly expressed throughout the Drosophila brain [4,35], including in the mushroom body, which is a key integration centre for sensory information that shares many architectural features with the vertebrate cerebellum [41]. The intrinsic neurons of the mushroom body, known as Kenyon cells, exhibit nuclear condensate formation and impaired long-term memory upon expression of either human or Drosophila HDAC4 [4,42]. Developmental overexpression of HDAC4 disrupts both mushroom body and eye formation [35,42,43], and the severity is exacerbated by nuclear accumulation of HDAC4 [35,42].
The propensity of HDAC4 to form condensates appears to stem from its glutamine-rich N-terminus, as deletion of residues 1−179 of human HDAC4 prevents nuclear condensate formation [37]. This region mediates homo-oligomerization of HDAC4 through its N-terminal α-helix; in solution, the N-terminus of human HDAC4 forms an α-helix that self-associates in a four-helix bundle [36]. This tetramerization is dependent on the formation of a small hydrophobic core, consisting of Leu89, Ile90 and Phe93, which is supported by glutamine-dominated polar interaction networks. Substitution of Phe93 for a polar residue abolishes tetramerization in solution, while mutation of nearby His97 to phenylalanine extends the core and stabilizes tetramerization [36]. Furthermore, it was recently demonstrated that in the presence of DNA and MEF2, the N-terminal region of HDAC4 preferentially forms a dimer, with each HDAC4 molecule bound to a MEF2 dimer that associates with its cognate DNA molecule [44].
*Mutation of the hydrophobic core or conserved glutamine residues alters oligomerization of DmHDAC4 in the adult brain. (A) Schematic diagram depicting the domain structure of Drosophila HDAC4 (top, DmHDAC4), including the N-terminal α-helix, MEF2 binding site (MEF2, purple) nuclear localization signal (NLS, green), 14-3-3 binding sites (P, phosphorylated serine (S) residues, yellow), catalytic domain (teal), nuclear export signal (NES, orange) and C-terminal Myc-tag (grey). Alignment of the α-helix region of hsHDAC4 and DmHDAC4 (bottom). Mutants used in this study: F65A, green box; 9QA, red boxes; 9QA∆Q, red boxes and grey ∆Q; S69F, orange box. In the 3SA mutant, the three phosphorylated serines (S) were substituted for alanines. (B) SDS-PAGE of chemically crosslinked purified recombinant wild-type and mutant N-terminal HDAC4 (HDAC4N). In the absence of crosslinker (–, DMSO only) HDAC4N appears as an approximately 15 kDa monomer (1). Addition of 0.2 mM or 2.0 mM disuccinimidyl suberate (DSS) induces crosslinking, enabling visualization of dimer (2), trimer (3) and tetramer (4) species. (C) Quantification of HDAC4N oligomerization following treatment with 2.0 mM DSS. The average proportion of monomer, dimer, trimer and tetramer species for each lane is displayed (n = 3 experiments). HDAC4N-F65A is significantly reduced in trimer (p = 0.03091) and tetramer (p = 0.009762), and increased in monomer species compared with HDAC4N-WT (p = 0.0002848). HDAC4N-S69F is significantly increased in trimer species compared with HDAC4N-WT (p = 0.0282). ANOVA: monomer F(2,6) = 59.77, p < 0.0001; dimer F(2,6) = 2.67, p = 0.1483; trimer F(2,6) = 24.57, p = 0.001288; tetramer F(2,6) = 15.51, p = 0.004256. Post hoc Tukey’s HSD, *p < 0.05, **p < 0.01, ***p < 0.001. Error bars indicate SEM. (D) Circular dichroism spectra of purified HDAC4N variants. HDAC4N-WT and HDAC4N-S69F exhibit α-helical folding, with characteristic negative peaks at 208 and 222 nm. HDAC4N-F65A shows a partial loss of α-helicity, while HDAC4N-9QA exhibits a more pronounced reduction in α-helicity, indicating a shift towards a less ordered conformation. (E) 222/208 nm ratios from CD spectra of purified HDAC4N. HDAC4N-WT and HDAC4N-S69F maintain a ratio close to 1, consistent with oligomerization, whereas HDAC4N-F65A shows a significant reduction, suggesting impaired oligomerization (n = 3 independent spectra). ANOVA, F(2,7) = 13.77, p = 0.003747; post hoc Tukey’s HSD, *p < 0.01. Error bars indicate SEM. (F) Whole cell lysates generated from adult heads expressing HDAC4WT-Myc or HDAC4Mutant-Myc under the control of elav-GAL4 were subjected to SDS-PAGE and probed for Myc and tubulin. The control is elav-GAL4/+. (G) Quantification of HDAC4 band intensity normalized to tubulin (n = 3 blots). HDAC49QA and HDAC49QA∆Q are significantly reduced compared with HDAC4WT. One sample t‐test compared with HDAC4WT: HDAC49QA t(2) = 4.8747, p = 0.0396; HDAC49QA∆Q t(2) = 9.1283, p = 0.0118. (H) Schematic of the co-immunoprecipitation workflow used in (I). 1. GFP- (green) and Myc-tagged (blue) HDAC4 are expressed in flies with elav-GAL4. 2. HDAC4 condensates comprise homo- and hetero-tetramers of GFP- and Myc-tagged HDAC4 in Kenyon cell nuclei. 3. GFP-HDAC4 is immunoprecipitated from whole cell lysates of adult fly heads and co-immunoprecipitated HDAC4-Myc (WT or mutant) is detected. (I) Genotypes were generated by crossing elav-GAL4 females to males carrying the indicated HDAC4 transgene. Flies were raised at 18°C until eclosion when adults were transferred to 22°C to increase transgene expression. Co-immunoprecipitation yield of HDAC4F65A (lane 4), HDAC49QA (lane 5), and HDAC49QA∆Q (lane 6) is significantly reduced compared with HDAC4WT (lane 3) upon immunoprecipitation of GFP-HDAC4.
While HDAC4 condensates have been recently documented [39], the mechanisms driving their formation and their relevance to neuronal dysfunction remain unclear. Here we aimed to determine whether HDAC4 oligomerization is required for condensate formation, and whether disrupting this process alters HDAC4-induced neurodevelopmental phenotypes in Drosophila. Using full-length HDAC4 mutants based on structural data, we demonstrate that oligomerization promotes nuclear import and condensate formation and determined that both oligomerization and MEF2 binding contributed to neurodevelopmental defects. Importantly, the level of HDAC4 overexpression resulting in these defects is modest at approximately 1.3-fold higher than endogenous levels [35], suggesting the phenotypes are not simply artefacts of high overexpression. This approach allows us to probe the molecular features that drive condensate formation and assess their functional relevance in vivo in a model with controlled and physiologically relevant expression levels. These findings offer insight into HDAC4 function, and how its dysregulation may contribute to neurological disease.
Results
Characterization of HDAC4 oligomerization mutants
2.1.
We first sought to examine whether the mechanism of oligomerization of Drosophila HDAC4 (DmHDAC4) is conserved with human HDAC4 (hsHDAC4). The sequence of the N-terminal α-helix of hsHDAC4 is highly conserved with DmHDAC4 (figure 1A). Notably, the three non-polar residues that form the hydrophobic core (Leu61, Ile62 and Phe65 in Drosophila) are all strictly conserved, as are nine of the glutamine residues that stabilize intra- and interhelical interactions [36]. As in vitro tetramerization of hsHDAC4 is destabilized upon substitution of Phe93 within the hydrophobic core [36], we generated the homologous mutation within DmHDAC4, Phe65Ala (HDAC4^F65A^). Either side of this core are the glutamine residues that form intra- and interhelical polar interaction networks. To investigate the importance of these networks for tetramer formation, the nine conserved glutamine residues were substituted with alanines (HDAC4^9QA^). While the N-termini of both hsHDAC4 and DmHDAC4 are glutamine-rich, DmHDAC4 has a significant polyglutamine stretch C-terminal to the nine conserved glutamines. Given the role that polyglutamine regions have in protein aggregation [45], as well as the roles of glutamines in the potentially stabilizing interaction networks of the HDAC4 tetramer [36], this 32 amino acid stretch (residues Gln112–Gln143) containing 25 glutamine residues was deleted alongside the 9QA substitutions (HDAC4^9QA∆Q^) to determine whether it confers additional stability to DmHDAC4 oligomers. Conversely, since hsHDAC4 tetramers are stabilized by extension of the hydrophobic core via substitution of His97 to Phe [36], we also introduced a hydrophobic Phe at the aligned position in Drosophila HDAC4 (HDAC4^S69F^).
In vitro oligomerization
2.1.1.
To characterize the capacity of these mutants to oligomerize, in vitro chemical crosslinking experiments were performed using purified recombinant N-terminal fragments spanning Pro37 to Gln143 of DmHDAC4, which corresponds to the region of hsHDAC4 examined by [36]. To avoid confusion with full-length HDAC4 constructs, these are denoted as HDAC4^N-mutant^. HDAC4^N-WT^ formed oligomers corresponding to homodimer, trimer and tetramers as well as higher order species (figure 1B). HDAC4^N-F65A^ showed a significant reduction in trimer and tetramer species and was increased in monomer species compared with HDAC4^N-WT^, whereas HDAC4^N-S69F^ exhibited a significant increase in trimers (figure 1C). HDAC4^N-9QA^ behaved markedly differently—expression in E. coli was notably inefficient, requiring approximately 100-fold more culture volume to achieve comparable yields to HDAC4^N-WT^, and significant degradation occurred during affinity and size-exclusion purification. Crosslinking and SDS-PAGE analysis revealed a smear rather than discrete oligomeric species, indicating these degradation products are capable of oligomerization. Changes in oligomerization between the mutants and wild-type were further supported by shifts in retention volume during size-exclusion chromatography (electronic supplementary material, figure S1), consistent with findings for human HDAC4 [44]. Collectively, these results show that disruption of the hydrophobic core in HDAC4^N-F65A^ decreases oligomer formation with no impact on protein stability, whereas mutation of the glutamine residues in HDAC4^N-9QA^ severely compromises protein stability and proper oligomer formation.
Circular dichroism analysis revealed that HDAC4^N-WT^ primarily adopted an α-helical secondary conformation in solution, similarly to hsHDAC4 [36], as did HDAC4^N-F65A^ and HDAC4^N-S69F^ (figure 1D). However, features of α-helix formation were lost for HDAC4^N-9QA^, likely due to its inherent instability and the high concentration of degradation products. Comparison of the 222/208 nm ratio for structures that form an α-helix provides insight into helical packing and oligomerization of proteins, where ratios close to or above 1 suggest coiled-coil or tightly packed helices, and lower values indicate more isolated helices or structural destabilization [46]. Both HDAC4^N-WT^ and HDAC4^N-S69F^ had ratios close to 1 (figure 1E), supporting that they exist as oligomeric helices, while the ratio for HDAC4^N-F65A^ was reduced, suggestive of forming more isolated helices. These data indicate that the substitution of Phe65 to Ala destabilizes oligomerization, whereas it is stabilized by the substitution of Ser69 to Phe. Moreover the glutamine residues are required for the correct α-helical folding of HDAC4.
In vivo oligomerization
2.1.2.
To characterize the ability of each of the full-length HDAC4 mutants to oligomerize in vivo, transgenic flies were generated carrying UAS-HDAC4^WT^ or each of the mutants (hereafter collectively referred to as HDAC4^Mutant^) for UAS/GAL4 regulated expression in the brain [47]. We adopted an overexpression system using the pan-neuronal elav-GAL4 driver [48,49] for phenotypic analysis. In a previous study, we compared endogenous and transgene HDAC4 via western blot [35]. Based on quantification of those data, we determined that HDAC4 levels are elevated by approximately 1.3-fold over endogenous levels, indicating modest overexpression. The expression levels of HDAC4^F65A^ and HDAC4^S96F^ were not significantly different from those of HDAC4^WT^. By contrast, HDAC4^9QA^ and HDAC4^9QA∆Q^ were expressed at approximately half the level of HDAC4^WT^ (figure 1F,G; p < 0.001). This reduction in protein level is consistent with the biochemical evidence showing instability of HDAC4^9QA^ and suggests that, in addition to mediating oligomerization and stabilizing the N-terminal helix of HDAC4, the N-terminal glutamine residues contribute to the proper folding and stability of full length HDAC4 in vivo.
To determine whether the HDAC4 mutants retained the capacity to oligomerize, co-immunoprecipitation (co-IP) was performed on whole head lysates from flies in which UAS-GFP-HDAC4^WT^ [42] was pan-neuronally co-expressed with either UAS-HDAC4^WT^-Myc or UAS-HDAC4^Mutant^-Myc via the elav-GAL4 driver (figure 1H). Immunoprecipitation of GFP-HDAC4^WT^ led to robust co-IP of HDAC4^WT^-Myc (figure 1I, lane 3), confirming oligomerization of wild-type DmHDAC4. HDAC4^F65A^, HDAC4^9QA^ and HDAC4^9QA∆Q^ showed a marked reduction in co-IP (figure 1G, lanes 4−6), revealing a severely compromised ability to oligomerize. However, given that HDAC4^9QA^ and HDAC4^9QA∆Q^ were expressed at approximately half that of HDAC4^WT^, it is unclear whether the reduction in co-IP efficiency is a result of lower abundance and/or a change in oligomerization.
For the S69F mutation, which enhanced crosslinking in vitro (figure 1C), co-IP with GFP-HDAC4^WT^ was similar to HDAC4^WT^. However, the amount of GFP-HDAC4^WT^ recovered by immunoprecipitation was consistently reduced in the presence of HDAC4^S69F^, leading to a modest increase in the ratio of co-IPed HDAC4^S69F^-Myc relative to the GFP-HDAC4^WT^ input (figure 1I, lane 7). This could suggest more efficient incorporation of HDAC4^S69F^ into oligomers; however, this should be interpreted cautiously as co-IP efficiency may also be influenced by complex stoichiometry involving additional binding partners or saturation effects.
Together, these findings indicate that both the hydrophobic core and glutamine-mediated polar interaction networks and/or α-helical folding are essential for N-terminal oligomerization of HDAC4 in vitro, and for the self-association of full-length HDAC4 in vivo. For clarity, we refer to this multimerization as oligomerization, although the precise stoichiometry of HDAC4 assemblies in vivo (e.g. dimers, trimers or higher-order species) has not yet been resolved.
Oligomerization of HDAC4 correlates with nuclear condensate formation in mushroom body neuronal nuclei
2.2.
We next examined whether the impaired ability of HDAC4 to oligomerize led to reduced nuclear condensate formation in vivo. At low expression levels, HDAC4 condensates are present but difficult to visualize reliably due to weaker signal intensity. Therefore, higher expression was necessary to achieve robust and consistent detection of condensates across cells, enabling accurate quantification. To that end, we drove expression of UAS-HDAC4-Myc with the stronger mushroom body driver OK107-GAL4 [48,50]. To ensure that transgene expression did not impact the development of these cells, expression was induced in the adult brain via the TARGET system in which GAL4 is repressed during development at 18°C by a temperature-sensitive mutant of GAL80 (GAL80ts). Once adult flies emerged, they were transferred to 30°C, at which temperature GAL80ts repression of GAL4 is relieved and HDAC4 transgene expression is permitted [51] (figure 2A). MEF2 was used as a counterstain to enhance the visibility of HDAC4 condensates as it is sequestered in HDAC4 nuclear foci [4,12,35,52]. HDAC4^WT^ was robustly expressed in adult Kenyon cells (figure 2B,C), and formed nuclear condensates that colocalized with MEF2 (figure 2E), as observed previously [4,35]. Expression of HDAC4^F65A^, HDAC4^9QA^ and HDAC4^9QA∆Q^ resulted in significantly fewer condensates than HDAC4^WT^, whereas HDAC4^S69F^ expression doubled the number of condensates (figure 2D). These data collectively demonstrate a positive correlation between the N-terminal α-helix’s ability to oligomerize and the formation of condensates in vivo. Although condensate formation was significantly decreased by HDAC4^F65A^, HDAC4^9QA^ and HDAC4^9QA∆Q^, it was not completely abolished, likely due to the incomplete loss of oligomerization in each mutant (figure 1B). Additionally, oligomerization with endogenous HDAC4, which we have previously shown can form condensates with transgene HDAC4^WT^ [35], may also contribute to the observed condensate formation.
*HDAC4 oligomerization capacity correlates with formation of nuclear condensates. All genotypes were generated by crossing tubP-GAL80ts;OK107-GAL4 females to males carrying the indicated UAS-HDAC4-Myc transgene or the w(CS10) control. Transgene expression was induced in adults by shifting flies from 18°C (GAL80ts active) to 30°C (GAL80ts inactive) after eclosion. Brains were dissected 72 h post-induction. (A) Schematic of adult-specific gene induction protocol. (B,C,E) Whole mount brains stained with anti-Myc (green) and anti-MEF2 (magenta). (B) Maximum projection showing HDAC4WT-Myc expression, which colocalizes with endogenous MEF2 in Kenyon cell nuclei. Asterisk labels the calyx and the boxed region is depicted in (C). Scale bar = 200 µm. (C) Single optical section (0.5 µm) through the calyx and Kenyon cell layer showing HDAC4WT colocalization with MEF2. Scale bar = 50 µm. (D) Quantification of nuclear condensates (colocalizing HDAC4/MEF2 puncta) per 1 µm section (as shown in C) (n = 4 brains per genotype, 10 sections per brain). ANOVA, F(4,15) = 36.88, p < 0.0001; post hoc Tukey’s HSD, *p < 0.05, **p < 0.01, **p < 0.001. Error bars indicate SEM. (E) Representative 0.5 µm sections showing HDAC4 condensates (arrowheads). Scale bar = 5 µm.
HDAC4 forms dynamic condensates in vivo
2.3.
Our data demonstrate that oligomerization via the N-terminus is required for nuclear condensation of HDAC4. However, it remained unclear whether other regions of the protein contribute to this process. To investigate this, we compared full-length HA-tagged HDAC4 (HDAC4^WT^) with a truncated N-terminal construct of HDAC4 comprising residues Met1 to Leu285 (HDAC4^M1-L285^). This region includes the MEF2 binding site to facilitate nuclear import, and one of the key 14-3-3 binding sites (Ser 239), which we mutated to alanine to prevent nuclear exit (figure 3A). When expressed in adult Kenyon cells under the same conditions as in figure 2, HDAC4^WT^ was observed in both cytoplasmic haloes and in the nucleus, where it formed condensates (figure 3B), whereas HDAC4^M1-L285^ localized exclusively to the nucleus, where it appeared more uniform and granular than HDAC4^WT^. This lack of condensation persisted even under constitutive expression with OK107-GAL4 and elav-GAL4 throughout development, which facilitates prolonged accumulation (figure 3C,D). These observations indicate that while the N-terminal region enables oligomerization, additional sequences within the C-terminus are necessary for condensate formation. To confirm that this was not a result of differing protein levels in the nucleus, we assessed HDAC4 levels by immunoblotting. While total HDAC4^WT^ levels were four times higher than HDAC4^M1-L285^ in whole head lysates (figure 3E,F), both had comparable levels in nuclear fractions (figure 3G,H), indicating that the inability of HDAC4^M1-L285^ to form condensates is not due to reduced nuclear concentration. These data also validate that condensate formation is not due to the presence of the Myc tag, and further we have also observed condensates with FLAG-tagged [4] and GFP-tagged [42] HDAC4 constructs.
*HDAC4 condensate formation requires regions downstream of the N-terminus and is dynamic. (A) Schematic diagram depicting the domain structure of full-length DmHDAC4 (top) and the truncated HDAC4M1-L285 (bottom). Ser239 is mutated to Ala in HDAC4M1-L285. (B–D) Representative 0.5 µm optical sections through the Kenyon cell layer stained with anti-Myc (green) and anti-MEF2 (magenta). Scale bar = 5 µm. Insets show individual nuclei. (B) Expression of UAS-HDAC4WT-HA or UAS-HDAC4M1-L285-HA driven by OK107-GAL4 and induced in adulthood with GAL80ts (30°C, 72 h). The control is tubulin-GAL80ts/+;OK107-GAL4/+. (C) Expression of UAS-HDAC4WT-HA or UAS-HDAC4M1-L285-HA driven by OK107-GAL4 at 25°C. (D) Expression of UAS-HDAC4WT-HA or UAS-HDAC4M1-L285-HA driven by elav-GAL4 at 25°C. (E) Western blot of HDAC4WT or HDAC4M1-L285 in whole cell lysates generated from adult heads expressing HDAC4WT or HDAC4M1-L285 under the control of elav-GAL4. The control (con) is elav-GAL4/+. (F) Quantification of HDAC4 band intensity (from E) normalized to tubulin (n = 3). HDAC4M1-L285 is significantly reduced compared with HDAC4WT. One sample t‐test, t(2) = 12.633, p = 0.0062. Error bars indicate SEM. (G) Subcellular fractionation and western blotting was performed on fly heads. Membranes were probed with anti-HA, as well as anti-lamin and anti-tubulin to assess fractionation efficacy. (H) Quantification of nuclear HDAC4 (from G), normalized to lamin (n = 2). There is no significant difference in nuclear abundance between HDAC4WT and HDAC4M1-L285. One sample t‐test, t(1) = 3.348, p = 0.185. Error bars indicate SEM. (I) Quantification of HDAC4WT nuclear condensation. The number of condensates (colocalizing puncta of HDAC4 and MEF2) per nucleus were counted and averaged for n ≥ 28 nuclei per section for n = 5 brains per genotype. ANOVA, F(2,12) = 14.67, p = 0.000599; post hoc Tukey’s HSD, *p < 0.01. Error bars indicate SEM. (J) Quantification of HDAC4WT condensate size. Condensate size was measured and averaged for n ≥ 257 condensates per section for n = 5 brains per genotype. Unpaired t‐test, t(8) = 2.315, p = 0.0493. Error bars indicate SEM. (K,L) Live imaging of whole mount brains. Genotypes were generated by crossing OK107-GAL4 females to UAS-HDAC4WT-GFP males, and raised at 18°C throughout development and adulthood. Differential interference contrast (DIC) microscopy was used to visualize nuclei (grey) and intrinsic GFP fluorescence was detected (green). (K) HDAC4WT-GFP condensates are mobile within nuclei. (L) Coalescence of two HDAC4WT-GFP condensates. Scale bar = 2 µm. (M) Anti-Myc (green) immunohistochemistry on whole brains stained with 1-fluoro-2,5-bis(3-carboxy-4-hydroxystyryl)benzene (FSB, magenta). Representative single optical sections (0.5 µm) through the Kenyon cell layer are shown. Genotypes were generated as described in (B,C). Scale bar = 5 µm.
We also observed that constitutive expression of HDAC4^WT^ using OK107-GAL4 resulted in significantly fewer condensates per nucleus compared with Kenyon cells where expression was restricted to the adult brain using tubulin-GAL80ts;OK107-GAL4 (figure 3I). However, the condensates in the constitutive expression conditions were larger than those resulting from tubulin-GAL80ts;OK107-GAL4 expression, which were smaller than the diffraction limit and therefore not quantified. Furthermore, expression with OK107-GAL4 resulted in larger condensates than elav-GAL4 (figure 3J), consistent with stronger expression driven by OK107-GAL4 in Kenyon cells [48]. These observations suggest that, over time, condensates undergo fusion or coalescence, resulting in fewer but larger structures. To explore the dynamics of these structures, we performed live-imaging of HDAC4^WT^-GFP in mushroom body neurons. HDAC4 condensates displayed dynamic movement (137 out of 174 were mobile during a 100 s period) (figure 3K). Although fusion events were infrequent (three observed during the 100 s period) they did occur (figure 3L); therefore, HDAC4 forms dynamic condensates in vivo that can coalesce under certain circumstances.
Finally, we examined colocalization of HDAC4 condensates with the Congo red derivative FSB (1-fluoro-2,5-bis(3-carboxy-4-hydroxystyryl)benzene) and thioflavin-T (THT), markers of β-sheet amyloid aggregation [53]. FSB and THT successfully detected the 42 amino acid Aβ fragment of human Amyloid Precursor Protein in Kenyon cells when expressed under the control of OK107-GAL4 (figure 3M; electronic supplementary material, figure S2). However, neither FSB nor THT colocalized with HDAC4^WT^ condensates, suggesting that they are not dominated by amyloid cross β-sheet structures.
These findings highlight that HDAC4 condensate formation depends on both N-terminal oligomerization and C-terminal protein domains. This condensation occurs in a dynamic manner, influenced by expression level and duration, and lacks amyloid-like characteristics, suggesting that HDAC4 oligomerization and accumulation may have significant biological relevance. We continue to refer to HDAC4 inclusions as biomolecular condensates because their formation requires N-terminal self-association, and they exhibit hallmarks consistent with liquid–liquid phase separation (LLPS). These hallmarks include concentration-dependent foci formation and the ability to form into larger structures over time, features commonly observed in phase-separated condensates [54]. Furthermore, the involvement of intrinsically disordered domains and multivalent interactions within HDAC4 is consistent with LLPS-mediated assembly.
Condensation of HDAC4 correlates with the severity of HDAC4 overexpression-induced neurodevelopmental defects
2.4.
Building on these findings, we next examined whether the capacity of each mutant to form condensates was linked to the severity of neurodevelopmental defects induced by HDAC4 in the mushroom body. The Drosophila mushroom body is a bilateral structure composed of bundled axons and thus is ideal for monitoring gross changes in axon morphogenesis. These axons project from Kenyon cell bodies in the posterior of the brain in a bundle termed the pedunculus before bifurcating based on their subtype (α/β, α′/β′ and γ) to form characteristic lobes (figure 4A) [55]. The cell adhesion molecule fasciclin II (FasII) is abundantly expressed in the α/β and γ lobes, allowing for clear visualization of these lobes when immunostained with anti-FasII [56]. Consistent with previous findings [35,42], expression of HDAC4^WT^-Myc with the pan-neuronal elav-GAL4 driver resulted in various mushroom body defects including premature lobe termination (missing lobe), lobe thinning and lobe fusion (figure 4A, table 1).
*HDAC4 oligomerization correlates with severity of defects in mushroom body development. (A) Immunohistochemistry with anti-fasciclin II (FasII) labels the alpha (α), beta (β) and gamma (γ) lobes of the adult mushroom body. Maximum projections were generated from stacks acquired at 1 µm increments. Defects (arrows) resulting from HDAC4-overexpression include missing lobes, α and β lobe thinning, guidance defects, mild β lobe fusion, and severe β lobe fusion. Scale bar = 50 µm. (B–D) Quantification of β lobe fusion resulting from HDAC4 overexpression. All genotypes were generated by crossing elav-GAL4 females to males carrying the indicated UAS-HDAC4 transgene or the w(CS10) control. Mild fusion was counted when less than half of the β lobe was fused, and severe when fusion was greater than half. Significance was determined using Fisher’s exact test, **p < 0.01, **p < 0.001. (B) The proportion of brains displaying β lobe fusion was significantly reduced for HDAC4F65A, HDAC49QA and HDAC49QA∆Q mutants compared with HDAC4WT. (C) At lower expression levels, the difference in severity between HDAC4WT and HDAC4S69F appeared larger but this was not statistically significant. (D) HDAC4WT-Myc and HDAC4WT-HA both induced β lobe fusion to a similar degree, while no fusion was observed for HDAC4M1-L285-HA.
Table 1.: HDAC4 oligomerization mutants reduce HDAC4-overexpression-induced defects in mushroom body development. All genotypes were generated by crossing elav-GAL4 females to males carrying each UAS-HDAC4-Myc transgene and to the w(CS10) control. The percentage of brains displaying each phenotype was calculated from the total number of brains analysed for each genotype (n). Statistical analysis was performed with Fisher’s exact test. Overexpression of HDAC4WT resulted in a significant number of brains with β lobe fusion compared with control (p < 0.0001). Expression of HDAC4F65A significantly reduced β lobe fusion compared with HDAC4WT (p = 0.0017), as did expression of HDAC49QA (p < 0.0001) and HDAC49QA∆Q (p < 0.0001). Percentages may not sum to 100 due to rounding.
The most prominent defect was β lobe fusion, which results from a failure of the β lobe axons to correctly terminate, resulting in erroneous crossing of the midline. β lobe fusion was observed in the majority of HDAC4^WT^ brains, but was significantly reduced in HDAC4^F65A^, HDAC4^9QA^ and HDAC4^9QA∆Q^ brains (figure 4B, table 1). Subclassification of fusion severity to either mild or severe revealed that these mutant forms also reduced the severity of the phenotype (table 1). It should be noted that as HDAC4^9QA^ and HDAC4^9QAΔQ^ protein levels were only half that of HDAC4^WT^ and HDAC4^F65A^, the decreased phenotype may be attributable to their lower expression levels, as we have previously observed that the severity of β lobe fusion correlates with the level of HDAC4 expression [35]. Since neither HDAC4^9QA^ nor HDAC4^F65A^ completely eliminated condensation or mushroom body defects, we combined HDAC4^9QA^ and HDAC4^F65A^ mutations as we hypothesized this may further destabilize oligomerization and subsequent HDAC4 condensation; however, HDAC4^9QA-F65A^ did not further reduce β lobe fusion (electronic supplementary material, table S1, figure S3).
Given the almost complete penetrance of β lobe fusion resulting from expression of HDAC4^WT^, it was unsurprising that expression of HDAC4^S69F^ did not result in a further increase. As β lobe fusion severity correlates with the level of expression [35], we reduced GAL4 activity by lowering the temperature [57], to detect more subtle differences. Under these conditions, β lobe fusion remained near complete in HDAC4^S69F^ brains (100% total, 72% severe), and was reduced for HDAC4^WT^ (84% total, 42% severe), although not significantly (p = 0.0687) (table 1, figure 4C). Nevertheless, together these data together demonstrate a correlation between nuclear condensation of HDAC4 and the severity of β lobe fusion in the mushroom body.
We also confirmed that the observed phenotypes are not dependent on epitope tag choice, as expression of HDAC4^WT^-HA mirrored the defects seen with HDAC4^WT^-Myc (table 2). By contrast, expression of the N-terminal HDAC4^M1–L285^ fragment, which includes the oligomerization domain but lacks the C-terminal region, did not induce any detectable mushroom body defects (table 2, figure 4D), indicating that while the N-terminal region is required for condensation, it is not sufficient to disrupt mushroom body development. These data therefore support a model in which nuclear condensation of full-length HDAC4 is associated with, and likely contributes to, its deleterious effects in the developing brain.
Table 2.: Full-length HDAC4 overexpression induces defects in mushroom body development. All genotypes were generated by crossing elav-GAL4 females to males carrying each UAS-HDAC4 transgene and to the w(CS10) control. Flies were raised at 22°C. The percentage of brains displaying each phenotype was calculated from the total number of brains analysed for each genotype (n). Statistical analysis was performed with Fisher’s exact test. Overexpression of HDAC4WT-Myc and HDAC4WT-HA each induced severe β lobe fusion (p < 0.0001).
Overexpression of HDAC4 is also associated with impaired eye development in Drosophila [35,42,43]. Development of the Drosophila eye initiates in early larval development and this process has been extensively characterized [58–60]. To investigate whether the defects in eye development similarly correlate with HDAC4 condensation, we used the glass multimer reporter driver (GMR-GAL4) to express HDAC4 transgenes in post-mitotic photoreceptors [61]. Expression of HDAC4^WT^ resulted in significant morphological abnormalities including fusion, and disorganization of ommatidia, loss of pigmentation, and the absence of bristles (figure 5A,B), consistent with previous findings [35,42,43]. Similar disruption was also observed for HDAC4^WT^-HA but not for HDAC4^M1-L285^-HA, indicating that the phenotype is dependent on the full-length protein, and independent of the epitope tag (electronic supplementary material, figure S4). HDAC4^F65A^ had no detectable effect on eye morphology (figure 5A–C), suggesting that disruption of oligomerization via this interface abrogates HDAC4’s ability to impair eye development. By contrast, HDAC4^9QA^ and HDAC4^9QA∆Q^ produced milder but still detectable eye defects, pointing to a more complex role for these mutants. Expression of HDAC4^S69F^ exacerbated the defects with almost complete fusion of all ommatidia and formation of cavities. To directly correlate changes in severity of the eye phenotype with condensation of HDAC4, immunohistochemistry was performed on the third instar larval eye disc (figure 5D), which represents the developmental window during which the adult eye is patterned and formed. As previously reported [35], HDAC4^WT^ formed prominent nuclear condensates in the undifferentiated basal cells of the disc (figure 5E,F). HDAC4^S69F^, which exacerbated eye defects, showed increased condensation. By contrast, HDAC4^F65A^, HDAC4^9QA^ and HDAC4^9QA∆Q^ showed markedly fewer and smaller nuclear condensates, often appearing as cytoplasmic haloes rather than concentrated nuclear foci (figure 5F).
*Oligomerization mutants alter HDAC4-overexpression-induced defects in eye development. (A) Stereomicrographs (110× magnification) and (B) scanning electron micrographs of adult Drosophila eyes of flies raised at 25°C. Flies carry one copy of GMR-GAL4 and two copies of the HDAC4-Myc wild-type or mutant transgene (GMR-GAL4/+; HDAC4/HDAC4). The control is GMR-GAL4/+; +. Scale bar = 50 µm. (C) Quantification of eye phenotype severity. HDAC4F65A, HDAC49QA and HDAC49QA∆Q mutants were significantly reduced, and HDAC4S69F was significantly increased in severity compared with HDAC4WT. ANOVA, F(5,112) = 1642.19, p < 0.001, post hoc Tukey’s HSD, **p < 0.001. Error bars indicate SEM. (D) Maximum projection of a third instar larval eye disc expressing HDAC4WT (green) and GFP.nls (magenta) under the control of GMR-GAL4. Flies were raised at 25°C. Expression of both HDAC4WT and GFP.nls was observed posterior to the morphogenetic furrow (arrowhead). DAPI (blue) highlights nuclei across the whole disc. The dashed line indicates the location of the cross section depicted in (E). Scale bar = 100 µm. (E) Cross section of the larval eye disc demonstrating that HDAC4WT localizes to both the apical and basal cells of the disc, but only forms small nuclear condensates (arrowheads) in undifferentiated basal cells. A photoreceptor precluster that forms a single ommatidium is circled in the apical surface, where HDAC4 condensates were not observed. Scale bar = 10 µm. (F) Single optical sections (0.5 µm) of basal undifferentiated cells of the larval eye disc expressing HDAC4WT or HDAC4Mutant-Myc (green), co-labelled with DAPI (blue). Scale bar = 10 µm.
Collectively these data further support a model in which HDAC4 condensation, mediated by self-oligomerization through the N-terminal α-helix, generally correlates with developmental defects in both the eye and mushroom body. However, this is not strictly linear—the differential phenotypes observed for HDAC4^F65A^ and HDAC4^9QA^ in the eye, despite both showing reduced condensation, highlight that additional factors such as protein stability, nuclear trafficking, or altered interactions with endogenous partners may modulate function in a tissue-specific manner. Thus, while nuclear condensation appears to be a key feature associated with HDAC4-induced pathology, the biological consequences of disrupting this process can differ depending on the nature of the mutation and the cellular context.
Both HDAC4 oligomerization and MEF2 regulate nuclear entry
2.5.
HDAC4 undergoes nucleocytoplasmic shuttling in response to stimuli [3,24–27], with subcellular distribution differing across cell types and species [9,42]. Despite intrinsic nuclear import and export signals, nuclear import of HDAC4 is primarily mediated through MEF2 binding, with point mutations that abrogate MEF2 binding resulting in cytoplasmic retention [14]. Previous studies showed that introducing the Phe93Asp mutation into hsHDAC4 (analogous to DmHDAC4^F65A^) impaired MEF2-dependent transcriptional repression [36,44] suggesting that HDAC4 oligomerization may be necessary for MEF2 binding and proper subcellular localization.
To explore this, we examined HDAC4 distribution in the Drosophila adult brain using elav-GAL4. Within Kenyon cells, HDAC4^WT^-Myc localized to the nucleus and cytoplasm with relatively uniform distribution and condensates were visible (figure 6A). Strikingly, HDAC4^F65A^, HDAC4^9QA^ and HDAC4^9QAΔQ^ displayed decreased nuclear localization and formed perinuclear haloes, whereas HDAC4^S96F^ fully localized to the nucleus where it formed prominent condensates.
Expression and subcellular localization of HDAC4 oligomerization mutants in the adult brain. All genotypes were generated by crossing elav-GAL4 females to males carrying the indicated UAS-HDAC4-Myc transgene. (A) Anti-Myc (green) and anti-MEF2 (magenta) immunohistochemistry was performed on whole brains. The schematic shows the Kenyon cell bodies viewed from the posterior, with the boxed region imaged via single optical 0.5 µm sections through the calyx and Kenyon cell layer. Arrowheads point to nuclear condensates of HDAC4. Scale bar = 10 µm. (B) Anti-Myc (green) and anti-MEF2 (magenta) immunohistochemistry and counterstaining with DAPI (blue) was performed on whole brains. The schematic shows the suboesophageal ganglion (SEG) and antennal lobe (AL) viewed from the anterior with the boxed region imaged via single optical 0.5 µm sections of the cells surrounding the AL and SEG. Scale bar = 10 µm.
By contrast, in cells surrounding the antennal lobes and the suboesophageal ganglion, HDAC4^WT^ was predominantly cytoplasmic, forming perinuclear haloes (figure 6B) and this distribution was unchanged for any of the mutants. Notably, these cells express a much lower level of MEF2 than Kenyon cells and MEF2 did not localize to cells in which HDAC4 was expressed. Together these findings indicate that HDAC4 subcellular distribution is cell type-specific, and its nuclear localization is closely associated with MEF2 expression, supporting a model in which oligomerization facilitates nuclear entry via MEF2 binding.
Both HDAC4 oligomerization and MEF2 binding contribute to HDAC4 condensation
2.6.
To specifically examine the role of oligomerization in condensate formation without the confounding effects of nuclear import, we generated HDAC4 3SA mutants by substituting serine residues at 14-3-3 binding sites with alanine (S239A, S573A, S748A), thereby blocking nuclear export [14,21,35]. These constructs allowed us to compare condensate formation across mutants with equivalent nuclear retention. Specifically, we introduced the F65A and 9QA mutations into the 3SA background to generate HDAC4^3SA-F65A^ and HDAC4^3SA-9QA^ , and also generated an HDAC4^3SA-F65A-ΔMEF2^ mutant to examine the contribution of MEF2 binding. Expression of all variants was confirmed via western blot (electronic supplementary material, figure S5A,B) and fractionation confirmed that the 3SA mutation increased their nuclear retention (electronic supplementary material, figure S5C–E), enabling unbiased comparison of nuclear condensation.
Condensate analysis revealed that HDAC4^3SA-F65A^ formed fewer condensates than HDAC4^3SA^ (figure 7A,B), indicating that oligomerization contributes to condensate formation. Disrupting both oligomerization and MEF2 binding via expression of HDAC4^3SA-F65A-ΔMEF2^ had an additive effect, resulting in almost complete abolishment of condensates. To confirm the role of MEF2 in condensate stabilization, we knocked down MEF2 via RNAi. This resulted in increased cytoplasmic localization of HDAC4^WT^ (figure 7C), consistent with MEF2-dependent nuclear import. By contrast, HDAC4^3SA^, which is restricted to the nucleus due to blocked export, remained nuclear but formed fewer condensates upon MEF2 knockdown (figure 7C,D). These results indicate that MEF2 also contributes to the stabilization of HDAC4 condensates in the nucleus.
*Functional analysis of nuclear HDAC4 oligomerization mutants and the role of MEF2. (A) Immunostaining of whole brains with anti-Myc (green) and anti-MEF2 (magenta). The indicated transgenes were driven with OK107-GAL4 and induced in adulthood with GAL80ts (30°C, 72 h). Representative 0.5 µm sections through the Kenyon cell layer are shown. Arrowheads point to nuclear condensates of HDAC4. Scale bar = 10 µm. (B) Quantification of nuclear condensates shown in (A). ANOVA, F(4,15) = 32.65, p < 0.0001; post hoc Tukey’s HSD, **p < 0.01, ***p < 0.001. Error bars indicate SEM. (C) Immunostaining of whole brains with anti-Myc (green) and anti-MEF2 (magenta). Flies were raised as in (A). Representative 0.5 µm sections through the Kenyon cell layer are shown. Arrowheads point to nuclear condensates of HDAC4. Scale bar = 10 µm. (D) Quantification of HDAC43SA condensates shown in (C). Condensates were counted and averaged for n ≥ 43 nuclei per section for n ≥ 3 brains per genotype. Unpaired t‐test, HDAC43SA versus HDAC43SA + MEF2 RNAi t(5) = 2.759, p = 0.0399. Error bars indicate SEM. (E,F) Quantification of β lobe fusion resulting from HDAC4 transgene expression. (E) Flies were raised at 22°C. HDAC43SA-9QA, HDAC43SA-∆MEF2 and HDAC43SA-F65A-∆MEF2 mutants all displayed a significant reduction in severe β lobe fusion compared with HDAC43SA, p = 0.0019, p < 0.0001 and p < 0.0001, respectively. *p < 0.05, **p < 0.01, ***p < 0.001, Fisher’s exact test. Error bars indicate SEM. (F) Flies were raised at 18°C. HDAC43SA-F65A mutants displayed a significant reduction in severe β lobe fusion compared with HDAC43SA, p = 0.0138. *p < 0.05, **p < 0.01, ***p < 0.001, Fisher’s exact test. Error bars indicate SEM. (G) Stereomicrographs (110× magnification) of adult Drosophila eyes of flies raised at 25°C. Flies carry one copy of GMR-GAL4 and two copies of the HDAC4Mutant-Myc transgene (GMR-GAL4/+; HDAC4/HDAC4). The control is GMR-GAL4/+; +. (H) Quantification of eye phenotype severity. Significant differences between HDAC43SA and HDAC43SA-Mutant were observed. Kruskal–Wallis test, H(5) = 106.92, p < 0.001, post hoc Dunn’s test, *p < 0.05, **p < 0.001. HDAC43SA versus HDAC43SA-9QA, p = 0.01561; versus HDAC43SA-F65A, p < 0.001; versus HDAC43SA-∆MEF2, p = 0.7861; versus HDAC43SA-F65A-∆MEF2 p < 0.001. HDAC43SA-F65A versus HDAC43SA-F65A-∆MEF2 p = 0.01387. Control versus HDAC43SA-F65A-∆MEF2 p = 0.3387. Error bars indicate SEM.
Oligomerization and MEF2 binding drive HDAC4-induced neurodevelopmental defects
2.7.
We next examined how these mutations affected mushroom body development. Expression of HDAC4^3SA^ caused severe β lobe fusion in 100% of brains, while HDAC4^3SA-9QA^ reduced this to 59% (table 3, figure 7E). HDAC4^3SA-F65A^ expression decreased severe β lobe fusion to 80%, which was not significantly different from HDAC4^3SA^. Strikingly, HDAC4^3SA-ΔMEF2^ and HDAC4^3SA-F65A-ΔMEF2^ mutations eliminated severe fusion, and total fusion was reduced to 30% and 11% respectively. Since HDAC4^3SA^ expression already caused maximal β lobe fusion, we lowered the temperature to reduce expression. Under these conditions, severe fusion was observed in 90% of HDAC4^3SA^ brains, but was reduced to 50% in HDAC4^3SA-F65A^ brains (figure 7F). Together these data demonstrate that mutations impairing HDAC4 oligomerization reduce β lobe fusion, independent of subcellular distribution.
Table 3.: Nuclear-restricted HDAC4 oligomerization mutants reduce HDAC4-overexpression-induced defects in mushroom body development. All genotypes were generated by crossing elav-GAL4 females to males carrying each UAS-HDAC4-Myc transgene and to the w(CS10) control. The percentage of brains displaying each phenotype was calculated from the total number of brains analysed for each genotype (n). At 22°C overexpression of HDAC43SA resulted in a significant number of brains with β lobe fusion compared with control (p < 0.0001, Fisher’s exact test). HDAC43SA-9QA, HDAC43SA-∆MEF2 and HDAC43SA-F65A-∆MEF2 mutants each significantly reduced severe β lobe fusion compared with HDAC43SA (p = 0.0019, p < 0.0001 and p < 0.0001, respectively). At 18°C severe β lobe fusion was significantly reduced for HDAC43SA-F65A compared with HDAC43SA (p = 0.0138). Severe β lobe fusion was reduced for HDAC4F65A compared with HDAC4WT (p < 0.0001). Percentages may not sum to 100 due to rounding.
In the eye, expression of HDAC4^3SA^ caused severe developmental disruption, including complete fusion of ommatidia, loss of pigmentation, loss of bristles and necrosis (figure 7G,H). Expression of HDAC4^3SA-9QA^ slightly reduced the severity with fewer necrotic spots, though fusion and loss of pigmentation remained. HDAC4^3SA-F65A^ provided clearer improvement, with some restoration of pigmentation and ommatidial structure. Notably, HDAC4^3SA-F65A-∆MEF2^ almost fully rescued the phenotype, producing eyes that appeared largely wild-type. By contrast, HDAC4^3SA-ΔMEF2^ showed only minor improvement, suggesting that oligomerization is essential for HDAC4-induced eye disruption. Since we previously illustrated that MEF2 binding is not required by HDAC4 to disrupt eye development, and detected no endogenous expression of MEF2 in the developing eye [35], these findings also suggest that the MEF2 binding site may facilitate interactions with an unidentified factor required for HDAC4-induced neurodevelopmental disruption in this tissue. Thus, both HDAC4 oligomerization and the MEF2 binding site are required for HDAC4-induced developmental disruption in the eye.
Discussion
Here we demonstrate that oligomerization of HDAC4 is essential for nuclear condensation and contributes to overexpression-induced neurodevelopmental defects in Drosophila. Using mutants that disrupt oligomerization and/or MEF2 binding, we dissected the contributions of these domains to condensate formation and neurotoxicity. Mutations in the N-terminal region that impaired oligomerization (HDAC4^F65A^ and HDAC4^9QA^) reduced condensate number in vivo, and this correlated with reduced severity of HDAC4-induced phenotypes in the mushroom body and adult eye, although this was not a linear relationship, as further discussed below. The relationship between nuclear condensates and phenotype severity was also somewhat confounded by the fact that these mutations also reduced the nuclear abundance of HDAC4. To resolve this, we enforced nuclear localization using a 3SA mutant background that disrupts 14-3-3-mediated export. Even when confined to the nucleus, HDAC4^3SA-F65A^ formed fewer condensates, confirming a direct role for oligomerization in condensate formation. Importantly, additional disruption of MEF2 binding further destabilized condensates and improved developmental outcomes, especially in the eye. These findings underscore a cooperative role for oligomerization and MEF2 binding in HDAC4 nuclear accumulation and subsequent condensate formation. Together, these findings establish HDAC4 oligomerization and MEF2 binding as key drivers of HDAC4-induced neurotoxicity, linking nuclear condensates to neurodevelopmental pathology.
HDAC4 is observed in small granular or punctate nuclear and cytoplasmic foci in neurons of both healthy mouse and Drosophila brains [9,35], suggesting that condensate formation may be a feature of its normal function. We hypothesize that small, dynamic HDAC4 condensates act as regulatory sites, potentially through interaction with transcription factors such as MEF2. Upon increased HDAC4 nuclear abundance, whether by overexpression, impaired import or unknown disease mechanisms, these puncta may increase in size and number, potentially reflecting fusion or coalescence events, and we hypothesize that this may induce neuronal dysfunction. While we observed such events for GFP-tagged HDAC4 under overexpression conditions, whether this occurs for endogenous HDAC4 remains to be investigated.
We determined that the mechanism of HDAC4 oligomerization is conserved between Drosophila and humans. In Drosophila, mutations that disrupt either the hydrophobic core or polar interaction networks in the N-terminus impair oligomerization in vitro and reduce condensation of full-length HDAC4 in vivo. These structural features are conserved across class IIa HDACs [36], suggesting oligomerization is critical to their function. Supporting this, HDAC4 oligomerization is required for repression of MEF2-dependent transcription in vitro [36,44]. In vivo, oligomerization-disrupting mutants led to increased cytoplasmic distribution of HDAC4 in Kenyon cells. This is likely due to impaired MEF2 binding, which is known to promote nuclear import [14,23] and increase condensation [35].
We also explored the role of glutamine-rich regions, which are frequently implicated in protein aggregation [62]. Mutating the glutamine residues implicated in oligomer stability (HDAC4^9QA^) reduced protein stability and in vivo condensation, and purified HDAC4^N-9QA^ was unstable in vitro. In cross-linking assays, the HDAC4^9QA^ mutant failed to form defined dimers or higher-order oligomers, instead producing a smeared profile suggestive of oligomerization of degradation products, or aberrant aggregation. This instability limited our ability to assess the precise contribution of this mutant to HDAC4 function. The decreased level of condensation observed in vivo was at least partially attributable to reduced protein levels rather than impaired oligomerization alone. Phenotypically, HDAC4^9QA^ induced milder mushroom body defects than HDAC4^WT^, consistent with its lower expression and inability to oligomerize. Unexpectedly, HDAC4^9QA^ produced an intermediate phenotype in the eye, while HDAC4^F65A^ had no phenotype. This suggests that the 9QA mutation, in addition to blocking oligomerization, may alter HDAC4 structure or interaction specificity in a way that affects eye development.
This structural complexity highlights the value of HDAC4^F65A^ as a more informative model for dissecting the role of oligomerization. Unlike HDAC4^9QA^, HDAC4^F65A^ maintained wild-type protein levels and preserved its α-helical conformation, yet exhibited reduced oligomerization and condensation. Correspondingly, HDAC4^F65A^-induced phenotypes were milder than HDAC4^WT^, particularly in the mushroom body, and absent in the eye. Thus HDAC4^F65A^ provided a cleaner experimental tool to examine the specific effects of reduced oligomerization without confounding structural instability.
Importantly, disruption of the MEF2-binding domain or knockdown of MEF2 reduced nuclear condensation independently of HDAC4 self-oligomerization and subcellular distribution, indicating that MEF2 not only facilitates nuclear localization but also stabilizes HDAC4 condensates. These findings raise the possibility that altered MEF2 expression could impair HDAC4 nucleocytoplasmic dynamics and contribute to its nuclear accumulation and condensation in neuronal disease. Further supporting a functional relationship between oligomerization and MEF2 binding, it has been shown that binding of MEF2’s HDAC-interacting domain to the N-terminus of human HDAC4 promotes dimerization and chromatin looping through the same hydrophobic core that mediates tetramerization [36,44]. This reinforces the idea that HDAC4 oligomerization underlies its chromatin regulatory functions.
Consistent with characteristics of LLPS, HDAC4 condensates were dose-dependent and dynamic. We show that HDAC4 nuclear condensation correlates with increased nuclear abundance, which has been observed in disease [28–30,33]. HDAC4 condensates are not β-sheet insoluble inclusions, but instead are dynamic and display features of LLPS in that their formation is both concentration- and interaction-dependent. Phase separation involves a nucleation event that seeds condensate formation [63], often involving a scaffold protein that recruits clients. HDAC4 oligomerization may constitute this nucleation trigger, with MEF2 acting as a client that further stabilizes condensation. Another key feature of LLPS is valency, which describes how the scaffold protein has many binding regions and motifs, including intrinsically disordered regions (IDRs) [40]. Indeed regions outside of the N-terminus of human HDAC4 are predicted to be intrinsically disordered [39] and are essential for Drosophila HDAC4 condensation, supporting a conserved role in phase separation.
Our data also point to a dominant negative effect of HDAC4 condensates. We previously demonstrated that endogenous HDAC4 is sequestered in condensates that result from GAL4-induced expression of transgene HDAC4 [35]. Here we show that oligomerization-reduced mutants still formed nuclear condensates when localized to the nucleus, likely supported by binding endogenous HDAC4. This could explain how heterozygous disease-associated mutations that disrupt 14-3-3 binding, which are predicted to promote nuclear retention [6], could contribute to pathology by driving condensation with wild-type HDAC4, potentially disrupting its normal function. These mutations have further significance in light of the recent finding that 14-3-3 binding to HDAC4 destabilizes condensate formation [39]. Further research is required to understand the relationship between the concentration of endogenous HDAC4 and condensate dynamics.
Tissue specific differences in HDAC4 function were evident. Although the MEF2 binding site is largely dispensable for HDAC4-induced disruption of eye development [35], its loss enhanced the phenotypic rescue conferred by HDAC4^3SA-F65A^. Despite the presence of MEF2 in the optic lobes of the brain [64], we have not been able to detect endogenous expression within developing photoreceptor nuclei [35], suggesting the MEF2 binding site of HDAC4 may mediate interactions with other critical factors. One possibility is serum response factor, which shares an overlapping binding site with MEF2 [14,65], and is implicated in the Drosophila eye development [66]. These findings highlight the need to consider cell-type-specific binding partners in understanding HDAC4 condensation and toxicity.
Conclusion
HDAC4 condensation is a dynamic process, dependent on oligomerization, dose, subcellular distribution and protein–protein interactions. We demonstrate a correlation between nuclear condensation of HDAC4 and neurodevelopmental defects in vivo, providing evidence that HDAC4 condensates contribute to neuronal dysfunction. MEF2 is a key partner that promotes both nuclear import and condensate stability, but additional interacting proteins likely contribute in a tissue-specific manner. These findings clarify the molecular basis of HDAC4 condensation, and highlight its potential as a therapeutic target for disorders in which HDAC4 regulation is disrupted.
Methods
Fly strains
5.1.
All flies were maintained on standard medium at 22°C with a 12 h light–dark cycle unless otherwise indicated. P{w[+mW.hs] = GawB}elav[c155] (elav-GAL4, BDSC 458), w[∗]; P{w[+mW.hs] = GawB}OK107 ey[OK107]/In(4)ci[D], ci[D] pan[ciD] sv[spa-pol] (OK107-GAL4, BDSC 854), w[∗ ]; P{w[+mC] = GAL4-ninaE.GMR}12 (GMR-GAL4, BDSC 1104), w[1118]; P{w[+mC] = UAS-GFP.nls}14 (UAS-GFP.nls, BDSC 4775) and P{w[+mC] = UAS-APP.Abeta42.B}m26a (UAS-Ab42, BDSC 33770) were obtained from the Bloomington Drosophila Stock Center. w∗; P{w+mC = tubP-GAL80ts}10; TM2/TM6B, Tb1 (tubP-GAL80ts) and w(CS10) strains were provided by R. Davis (Scripps Research Institute, Jupiter, FL). P{GD5039}v15550 (UAS-MEF2-RNAi, VDRC 15550) was obtained from the Vienna Drosophila Resource Center. GFP-HDAC4^WT^ was previously generated [42]. Homozygous lines carrying OK107-GAL4, tubP-GAL80ts and both GFP-HDAC4^WT^ and HDAC4^Mutant^-Myc were generated by standard genetic crosses. The open reading frame of wild-type DmHDAC4 was previously synthesized (nucleotides 461−4216, NCBI NM_132640) with a C-terminal 6× Myc tag [42], HDAC4^WT^-Myc, and mutagenesis performed by GenScript (New Jersey, USA) to obtain HDAC4^F65A^, HDAC4^9QA^, HDAC4^9QA∆Q^, HDAC4^S69F^, HDAC4^3SA-9QA^, HDAC4^3SA-F65A^, HDAC4^3SA-F65A-∆MEF2^, as well as HDAC4^3SA^ and HDAC4^3SA-∆MEF2^ as previously described [42]. Replacement of the HDAC4^WT^ 6× Myc tag with a 3× HA tag was carried out by Genscript. HDAC4^M1-L285^-HA (nucleotides 461–1315, NCBI NM_132640), with the Ser239Ala mutation was synthesized with a C-terminal 3× HA tag. HDAC4 constructs were cloned into the pUASTattB vector for germline transformation of Drosophila (GenetiVision, Houston, TX) using the P2 docking site at (3L)68A4. All strains were outcrossed for a minimum of five generations into the w(CS10) genetic background.
Protein expression, purification and biochemical analyses
5.2.
Protein sequence alignment of Drosophila (nucleotides 461–4216, NCBI NM_132640, translated) and human (NCBI NP_006028) HDAC4 was performed using EMBOSS Water pairwise sequence alignment (Madeira, Madhusoodanan [67], https://www.ebi.ac.uk/jdispatcher/psa/emboss_water, RRID:SCR_025141). Residues Ala62-Gln153 of hsHDAC4 were previously examined for tetramer formation [36] and the corresponding region of DmHDAC4 was determined as Pro37-Gln143. This region was codon optimized for E. coli and synthesized by GenScript to create HDAC4^N-WT^. This was followed by mutagenesis to create HDAC4^N-F65A^, HDAC4^N-9QA^ and HDAC4^N-S69F^. HDAC4^N^ constructs were cloned into the pET-15B expression vector in frame with the N-terminal 6× His tag. HDAC4^N^ were transformed into BL21(DE3) E. coli for recombinant protein expression. Cells were grown at 30°C in 2× YT culture medium containing ampicillin (100 µg ml^−1^) until OD_550_ reached approximately 0.6, at which time expression was induced with 0.5 mM IPTG and cells were grown at 20°C overnight (approx. 14 h, HDAC4^WT^ and HDAC4^S69F^, 1 l total culture volume) or at 25°C for 3 h (HDAC4^F65A^ and HDAC4^9QA^, 10 l total culture volume). Cells were harvested by centrifugation at 5000 × g for 10 min and pellets stored at −80°C. Pellets were thawed and resuspended in 20 ml (per litre of culture) lysis buffer (30 mM HEPES, pH 7.5, 300 mM NaCl, 10 µg ml^−1^ Pepstatin A, 1× cOmplete EDTA-free protease inhibitors (Roche)) before lysis via Cell Disruptor (Constant Systems) at 10 kpsi and sonication for 2 × 30 s pulses at 20 W. The lysate was clarified by centrifugation at 20 000 × g for 30 min before filter sterilization. The supernatant was subjected to nickel-affinity chromatography using Profinity IMAC Resin (BioRad) in IMAC buffer (30 mM HEPES, pH 7.5, 300 mM NaCl) with a gradient elution from 30 mM to 500 mM imidazole. A_215_ and A_230_ were examined to determine the fractions in which HDAC4^N^ eluted, and these were also run on SDS-PAGE to confirm HDAC4^N^ presence given the low number of tyrosine and tryptophan residues. Pooled fractions containing HDAC4^N^ were subjected to size exclusion chromatography on a Superdex 75 10/300 GL column (Cytiva) in SEC buffer (30 mM HEPES, pH 7.6, 150 mM NaCl), and fractions containing HDAC4^N^ were pooled and concentrated to approximately 0.5−2 mg ml^−1^ before snap freezing and storing at −80°C.
Crosslinking was performed as per Guo et al. [36]. Briefly, 0.5 mg ml^−1^ HDAC4^N^ diluted in SEC buffer (20 µl) was incubated with 2 µl disuccinimidyl suberate (DSS, Thermo Fisher Scientific) dissolved in DMSO for 30 min at room temperature. The reaction was quenched for 15 min with 1 µl of 1 M Tris (pH 7.5) before 5 µl was prepared for SDS-PAGE followed by colloidal Coomassie staining. Quantification was performed using the Gel Analyser plugin within ImageJ [68] (https://imagej.net/, RRID:SCR_003070). Proportional intensity was calculated and data transformed using centred log-ratio before performing a one-way ANOVA with post hoc Tukey’s HSD test with significance set at α = 0.05.
Circular dichroism was performed on HDAC4^N^ prepared in SEC buffer at 0.5 mg ml^−1^ in a 0.1 mm quartz cuvette. Spectra were recorded using a Chirascan spectrophotometer (Applied Photophysics). For each sample spectra were collected between 180 and 260 nm in 1 nm steps and averaged, and data were baseline corrected for the sample buffer.
SDS-PAGE and western blotting
5.3.
Whole cell lysates were produced by homogenizing snap-frozen Drosophila heads in either IP buffer (50 mM sodium chloride, 30 mM sodium pyrophosphate, 50 mM sodium fluoride, 10% glycerol, 0.5% Triton X-100, 0.5 mM PMSF, 25 mM Tris, pH 7.05, 1× protease inhibitors (cOmplete EDTA free Protease Inhibitor Cocktail (Roche)) or RIPA buffer (150 mM sodium chloride, 0.1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris pH 8.0, 1× protease inhibitors) and collecting the supernatant following centrifugation at 12 000 × g for 2 min. Nuclear and cytoplasmic lysates were prepared using the NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific) using a protocol modified from Maitra et al. [69]. Heads (n = 50) were homogenized in 100 µl of cytoplasmic buffer 1 (CB1) supplemented with protease inhibitors and incubated on ice for 10 mins before adding 5.5 µl of cytoplasmic buffer 2 (CB2) and centrifugation at 10 000 × g for 5 min. The supernatant was retained as the cytoplasmic fraction, and the pellet washed twice by resuspending in 50 µl of CB1, incubating for 10 min, then adding 2.75 µl of CB2, centrifuging at 10 000 × g for 5 min and discarding the supernatant. The pellet was resuspended in 50 µl of nuclear extraction reagent with protease inhibitors, incubated on ice for 40 min and centrifuged at 10 000 × g for 10 min before collecting the supernatant as the nuclear fraction. Total protein was quantified using a Pierce BCA Protein Assay kit (Thermo Fisher Scientific). Whole cell lysate (30 µg) or subcellular fractions (20 µg) were denatured in 1× sample buffer (2% SDS, 5% 2-mercaptoethanol, 10% glycerol, 0.01% bromophenol blue, 60 mM Tris HCl, pH 6.8) at 90°C for 5 min. Samples were loaded onto 4–20% or 10% Mini-PROTEAN TGX Precast Protein gels (BioRad) and electrophoresed at 200 V. Protein was blotted onto a nitrocellulose membrane for 1 h at 4°C before blocking in 5% skim milk powder in TBST (20 mM Tris, 150 mM NaCl, pH 7.6, 0.1% Tween-20) for 1 h at room temperature. Following washing in TBST membranes were incubated overnight at 4°C in primary antibody in 1% skim milk powder/TBST, and 1 h at room temperature in secondary antibody. Antibodies used were rabbit anti-Myc (ab9106, Abcam, 1:1000, Antibody Registry Identifier RRID:AB_307014), rabbit anti-GFP (ab290, Abcam, 1:4000, RRID:AB_303395), rat anti-HA (Roche, clone 3F10, 1:1000, RRID:AB_2687407), mouse anti-α-tubulin (12G10 clone, Developmental Studies Hybridoma Bank (DSHB), 1:500, RRID:AB_1157911), mouse anti-lamin(Dm0) (ADL67.10 clone, DSHB, RRID:AB_528336), sheep anti-mouse-HRP (Sigma Aldrich NA931VS, 1:20 000), donkey anti-rabbit-HRP (Sigma Aldrich NA934VS, 1:40 000), goat anti-rat-HRP (Abcam ab97057, 1:10 000, RRID:AB_10680316). Amersham ECL Prime detection reagent (GE Life Sciences) was used as per manufacturer’s instructions, and chemiluminescence detected using the Azure Biosystems C600 imaging system. Quantification of western blots was performed using the Gel Analyser plugin within ImageJ [68] (https://imagej.net/, RRID:SCR_003070). Whole cell and cytoplasmic lysate signal were normalized to tubulin, and nuclear lysate to lamin. For blots of whole cell lysates, the fold-change of the normalized signal was calculated relative to HDAC4^WT^. For blots from subcellular fractionation, the nuclear to cytoplasmic ratio was calculated for each sample and normalized to HDAC4^WT^.
Immunoprecipitation
5.4.
Protein A/G magnetic beads (Thermo Fisher Scientific, 25 µl) were resuspended and washed in TBS-T (20 mM Tris, 150 mM NaCl + 0.05% Tween-202) before incubation with 2 µl anti-GFP (ab290, Abcam, RRID:AB_303395) for 1 h at room temperature. Whole cell lysate (2 mg) was added to the antibody–bead mixture and incubated overnight at 4°C. Beads were washed three times in TBS-T and proteins eluted in 1× sample buffer for 10 min at room temperature. The supernatant was collected and incubated at 95°C for 5 min. Total eluate was loaded for SDS-PAGE and western blotting.
Immunohistochemistry
5.5.
Whole flies were prefixed in PFAT-DMSO (4% paraformaldehyde, PBS, 0.1% Triton X-100, 5% DMSO) for 1 h at room temperature, followed by washing and dissection in PBT (10 mM phosphate buffer, pH 7.4, 0.5% Triton X-100). For mushroom body analyses, adult flies were anaesthetized with CO_2_ and placed on ice prior to dissection. Third instar crawling larvae were washed and dissected in PBS (10 mM phosphate buffer, pH 7.4). Tissues were post-fixed in PFAT-DMSO for 20 min and stored in 100% methanol. Tissues were rehydrated in 50% methanol/PBT, blocked for 3 h at room temperature in immunobuffer (5% normal goat serum in PBT) and incubated overnight at room temperature with primary antibody (mouse anti-FasII, 1:20, DSHB clone 1D4, RRID:AB_528235; rabbit anti-GFP, 1:20 000, Abcam ab290, RRID:AB_303395; rat anti-HA, 1:500, Roche clone 3F10, RRID:AB_2687407; rabbit anti-MEF2, 1:500, gift from Bruce Paterson, National Cancer Institute, Bethesda; rabbit anti-Myc, 1:100, Abcam ab9106, RRID:AB_307014; mouse anti-Myc, 1:100, DSHB clone 9E10, RRID:AB_2266850), followed by overnight incubation at 4°C with secondary antibody (goat anti-mouse Alexa 555, 1:500, Invitrogen A-21422, RRID:AB_2535844; goat anti-rabbit Alexa 647, 1:500, Invitrogen A21244, RRID:AB_2535812; goat anti-rat Alexa 555, 1:500, Invitrogen A21434, RRID:AB_2535855), then washing in PBT. Brains were incubated with FSB (0.01% in 50% ethanol) or THT (0.25% in 50% ethanol), washed in 50% ethanol, and then washed further in PBT. Samples that required nuclear staining were washed in PBT before incubation with DAPI (1:20 000 in PBS) and all tissues were mounted in Antifade (90% glycerol, 0.2% n-propyl gallate, 10 mM phosphate buffer, pH 7.4 (Sigma Aldrich), 0.5% Triton X-100). Images were captured using a Leica TCS SP5 DM6000B confocal microscope or a Zeiss LSM900 super-resolution microscope, and processed using ImageJ software.
Live imaging
5.6.
Live imaging of adult Drosophila brains was performed using a Zeiss LSM900 super-resolution microscope using a protocol modified from [70]. Brains were dissected from live flies in PBS and immediately mounted in Voltalef 10S oil, a coverslip placed atop, and imaged using a 63× oil immersion lens.
Light microscopy
5.7.
Light microscopy was performed using an Olympus SZX16 stereo zoom microscope and CellSens Dimension (Olympus) imaging software. Flies were frozen at −20°C before thawing and imaging at 110× magnification. Constant light intensity and exposure was used. Images were imported into Adobe Photoshop (RRID:SCR_014199), Z-axis drift accounted for using the Auto-Align Layers function, and optical sections stacked using the Auto-Blend Layers function.
Scanning electron microscopy
5.8.
Scanning electron microscopy was performed as previously described [42]. Briefly, adult flies were anaesthetized and immersed in primary modified Karnovsky’s fixative (3% glutaraldehyde, 2% formaldehyde, 0.1 M sodium phosphate buffer and Triton X-100), followed by vacuum infiltration, ethanol dehydration and critical point drying. Heads were mounted onto aluminium stubs, sputter coated with gold (Bal-Tec SCD 050 sputter coater), and imaged using an FEI Quanta 200 environmental scanning electron microscope (20.00 kV, spot 4.0 nm).
Quantification of condensation and neurodevelopmental phenotypes
5.9.
HDAC4 and MEF2 condensates in Kenyon cell nuclei were counted using the Cell Counter plugin (https://imagej.net/ij/plugins/cell-counter.html, RRID:SCR_025376) in ImageJ and were only counted if they were visible in both single and merged channels. For panels in figures 2D and 7B and electronic supplementary material, figure S3, condensates were counted through ten serial optical sections at 1 µm increments, beginning at the first emergence of the calyx (n = 4 brains per genotype), imaged at 150× magnification. When condensates were present in adjacent slices they were only counted once. For panels in figures 3I and 7D, condensates per nucleus were counted in a single optical section (705× magnification) for ≥27 cells per section (n ≥ 3 brains per genotype) using QuPath [71] (https://qupath.github.io/, RRID:SCR_018257). Statistical significance was assessed using a one-way ANOVA with post hoc Tukey’s HSD test with significance set at α = 0.05. Condensate size was measured using QuPath, where only condensates larger than the diameter of the Airy disc (0.288 µm) were included. Statistical significance was assessed using a two-sample unpaired t‐test with significance set at α = 0.05. Condensate movement was assessed by generating maximum projection images from 50 time-lapse frames obtained over 100 s. Condensates were scored as moving if the projection lacked a single, well-defined spherical condensate, indicating blurring due to motion. Movement was confirmed by examining individual frames throughout the time series.
Assessment of mushroom body phenotypes was performed by a blinded scorer as per Tan et al. [35]. Z-stacks of mushroom body lobes were scored for the presence or absence of developmental defects in the α- and β lobes, including lobe thinning, absence and guidance defects, as well as β lobe fusion. Fusion was scored as mild if axons measuring less than half the width of the lobe crossed the midline of the brain, or as severe if axons measuring more than half the width of the lobe crossed the midline. Statistical significance was assessed using Fisher’s exact test.
Assessment of eye development was performed by a blinded scorer as per Tan et al. [35]. A scoring system was developed to assess severity of defects in morphology which considered three key facets of eye development that can be reproducibly analysed; bristle formation, ommatidia alignment, fusion and pigmentation. Each aspect was scored from 0 to 4, for a total severity score of 12 per eye, which was converted to % severity. Bristles: 0 = All bristles present and correctly placed, 1 = Most bristles correctly placed, with no more than a small number missing or extra, 2 = Moderate loss of bristles or misplaced bristles and/or large loss of bristles at edges of the eye, 3 = Few bristles, 4 = Complete absence of bristles. Ommatidia: 0 = Ommatidia correctly aligned and no fusion, 1 = Alignment of ommatidia altered but no fusion, 2 = Ommatidia alignment perturbed, altered depth of ommatidia boundary but no fusion, 3 = Ommatidia alignment perturbed and fusion, 4 = Complete absence of ommatidia (total fusion). Pigmentation: 0 = No changes in pigmentation, 1 = Small number of ommatidia missing pigment, 2 = Moderate loss of pigmentation across the eye, 3 = Pigmentation only around the edge of the eye, or in small spots across eye, 4 = No pigmentation. Where data were normally distributed statistical significance was assessed using a one-way ANOVA with post hoc Tukey’s HSD test with significance set at α = 0.05. Where data were not normally distributed, the Kruskal–Wallis test was used with significance set at α = 0.05, followed by a post hoc Dunn’s test with a Bonferroni corrected α.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wang WH, Cheng LC, Pan FY, Xue B, Wang D, Chen Z, Li C. 2011 Intracellular trafficking of histone deacetylase 4 regulates long‐term memory formation. Anat. Rec. 294, 1025–1034. (10.1002/ar.21389)21542139 · doi ↗ · pubmed ↗
- 2Kim MS, Akhtar MW, Adachi M, Mahgoub M, Bassel-Duby R, Kavalali ET, Olson EN, Monteggia LM. 2012 An essential role for histone deacetylase 4 in synaptic plasticity and memory formation. J. Neurosci. 32, 10879–10886. (10.1523/jneurosci.2089-12.2012)22875922 PMC 3480333 · doi ↗ · pubmed ↗
- 3Sando R, Gounko N, Pieraut S, Liao L, Yates J, Maximov A. 2012 HDAC 4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 151, 821–834. (10.1016/j.cell.2012.09.037)23141539 PMC 3496186 · doi ↗ · pubmed ↗
- 4Fitzsimons HL, Schwartz S, Given FM, Scott MJ. 2013 The histone deacetylase HDAC 4 regulates long-term memory in Drosophila. P Lo S One 8, e 83903. (10.1371/journal.pone.0083903)24349558 PMC 3857321 · doi ↗ · pubmed ↗
- 5Zhu Y et al. 2019 Class I Ia HDA Cs regulate learning and memory through dynamic experience-dependent repression of transcription. Nat. Commun. 10, 3469. (10.1038/s 41467-019-11409-0)31375688 PMC 6677776 · doi ↗ · pubmed ↗
- 6Wakeling E et al. 2021 Missense substitutions at a conserved 14-3-3 binding site in HDAC 4 cause a novel intellectual disability syndrome. Hum. Genet. Genom. Adv. 2, 100015. (10.1016/j.xhgg.2020.100015)PMC 784152733537682 · doi ↗ · pubmed ↗
- 7Trazzi S et al. 2016 HDAC 4: a key factor underlying brain developmental alterations in CDKL 5 disorder. Hum. Mol. Genet. 25, 3887–3907. (10.1093/hmg/ddw 231)27466189 · doi ↗ · pubmed ↗
- 8Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow C. 2007 Distribution of histone deacetylases 1–11 in the rat brain. J. Mol. Neurosci. 31, 47–58. (10.1007/BF 02686117)17416969 · doi ↗ · pubmed ↗