A Cryopreservation and Regeneration Protocol for Embryogenic Callus of Larix olgensis
Chen Wang, Wenna Zhao, Yu Liu, Hao Dong, Yajing Ning, Chengpeng Cui, Hanguo Zhang, Meng Li, Shujuan Li

TL;DR
This paper presents a successful method to cryopreserve and regenerate embryogenic callus in Larix olgensis, preserving its ability to grow into new plants.
Contribution
A novel and effective cryopreservation protocol for L. olgensis embryogenic callus that maintains embryogenic potential and regenerative capacity.
Findings
A 24-hour preculture with specific sucrose concentrations and cryoprotectants achieved a 100% recovery rate of cryopreserved embryogenic callus.
Cryopreserved callus successfully underwent somatic embryogenesis, germination, and rooting.
The protocol is suitable for long-term storage as cryopreservation duration did not affect cell viability or proliferation.
Abstract
Larix olgensis is a valuable timber species in northern China, typically propagated through somatic embryogenesis (SE). However, long-term subculture can lead to a loss of embryogenic potential. This study aimed to establish a simple and stable protocol for the cryopreservation and regeneration of L. olgensis embryogenic callus (EC) that preserves its SE potential and regenerative capacity. The slow-freezing method was employed for cryopreservation. A cryopreservation protocol for L. olgensis EC was developed by optimizing the preculture duration and conditions, cryoprotectant composition and thawing temperature. The results showed that optimal outcomes were achieved using a 24 h stepwise preculture on medium containing 0.2 and 0.4 mol∙L−1 sucrose, followed by cryoprotectant treatment with 0.4 mol∙L−1 sucrose, 2.5% (v/v) dimethyl sulfoxide (DMSO) and 10% polyethylene glycol 6000…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
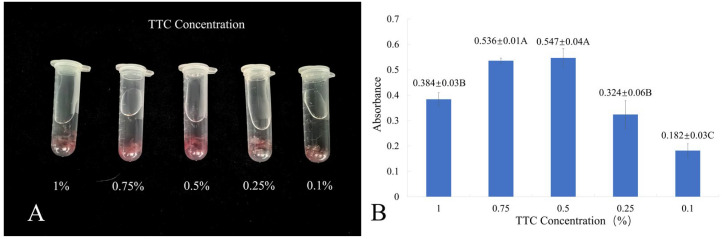 Figure 1
Figure 1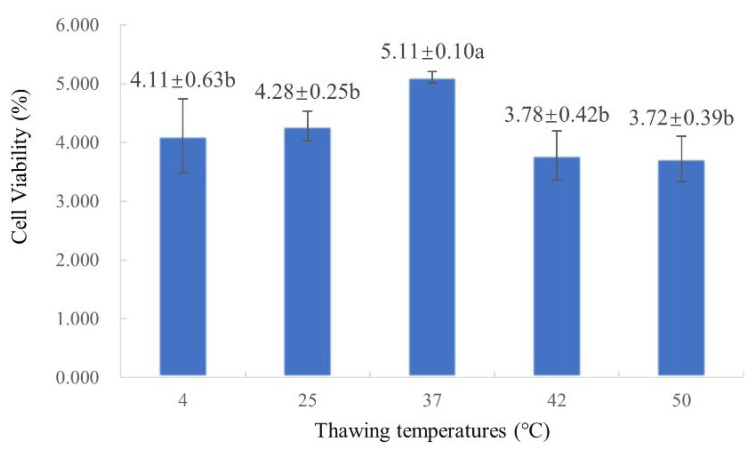 Figure 2
Figure 2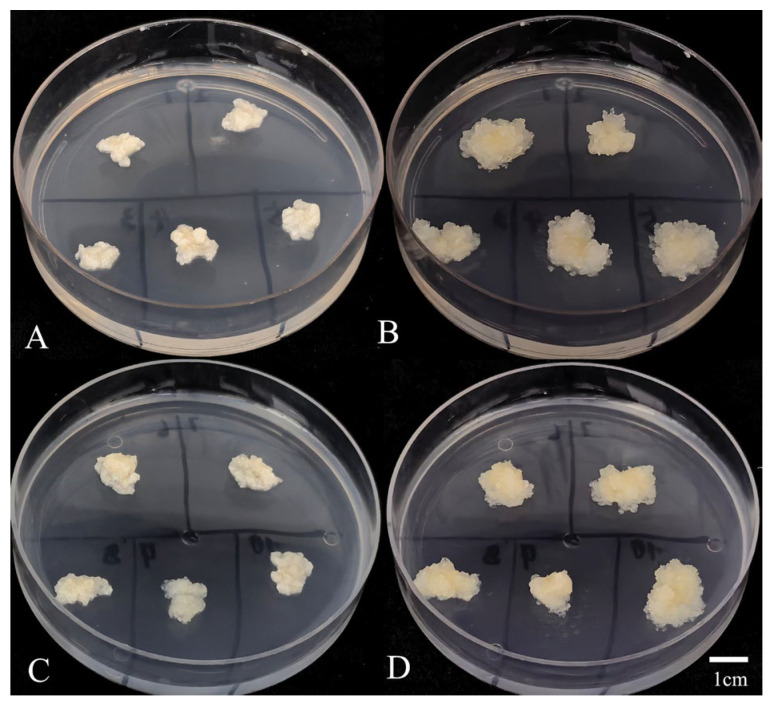 Figure 3
Figure 3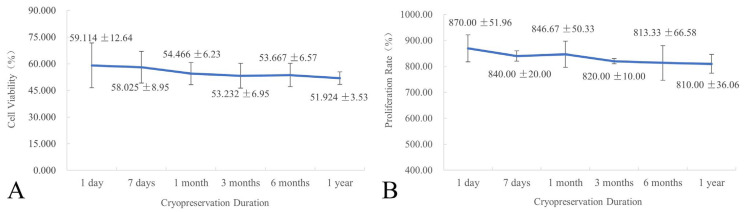 Figure 4
Figure 4 Figure 5
Figure 5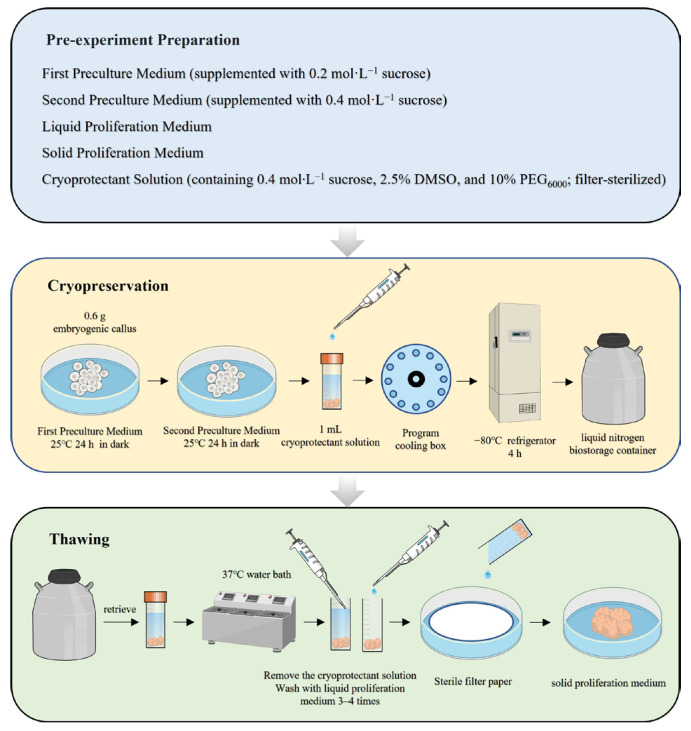 Figure 6
Figure 6- —Biological Breeding-National Science and Technology Major Projec
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInsect and Arachnid Ecology and Behavior · Plant Reproductive Biology · Seed Germination and Physiology
1. Introduction
Larix olgensis is a commercially valuable timber species in northern China and also possesses significant ecological importance. It contributes to soil and water conservation, air purification, and noise reduction and provides habitats for wildlife, playing an irreplaceable role in maintaining ecological balance [1]. Somatic embryogenesis (SE) is a process in which structures similar to zygotic embryos are formed directly from somatic cells without gamete fusion [2]. Compared to conventional breeding methods, SE offers several advantages, including high propagation efficiency, enhanced genetic stability, shorter breeding cycles, and lower production costs [3]. Furthermore, SE is highly controllable and easy to observe, making it valuable for both theoretical and applied research [4]. To date, SE has been successfully applied in L. olgensis for rapid propagation, germplasm preservation, and genetic improvement [5]. However, the long-term subculture of embryogenic callus (EC) leads to a series of adverse effects on subsequent development. These effects manifest as reduced cell viability, loss of regenerative capacity, alterations in gene expression patterns, and morphological and physiological abnormalities in regenerated plantlets [6]. These issues significantly impair the efficiency and stability of tissue regeneration. Cryopreservation refers to the long-term preservation of plant organs, tissues, or cells at the ultra-low temperature of −196 °C (typically in liquid nitrogen) [7]. At this temperature, EC are in metabolic standstill or suspended animation, in the case of the kinetic energy, enzymatic reactions, and molecular motion within biological systems, diminishing or ceasing entirely [8]. This technique is commonly integrated with in vitro culture methods. Therefore, the development of successful micropropagation systems is critical for cryopreserving propagules [9,10]. This enables the long-term, secure preservation of diverse materials, including pollens, calli, somatic embryos, zygotic embryos, seeds, shoot tips, and dormant buds [11,12,13,14]. Consequently, cryopreservation is universally recognized as the most effective and ideal method for the long-term preservation of plant germplasm resources [15]. The integration of SE and cryopreservation technologies shortens forest tree breeding cycles by allowing long-term cryopreservation of EC from diverse genotypes in liquid nitrogen. This approach minimizes the risks of somaclonal variation and reduces the labor and resources required for repeated subculturing [16]. Concurrently, elite genotypes identified through clonal field trials can be retrieved from cryopreservation, thawed, and mass-propagated via SE. This enables the rapid deployment of somatic embryo-derived plantlets in clonal forestry operations [17].
Since its initial development, cryopreservation protocols have been continuously refined. Current methods can be broadly classified into three categories: conventional techniques such as rapid-freezing [18] and slow-freezing [19] (used for over 40 years); vitrification-based approaches, including droplet vitrification [20], encapsulation–vitrification [21], and standard vitrification [22] and alternative methods such as air desiccation-freezing [23] and cryoplate protocols [24,25]. Each method has distinct advantages and limitations, and their suitability varies across plant species. Among these, conventional slow-freezing and vitrification-based methods are the most widely used in plant cryopreservation, with slow-freezing being particularly common in coniferous species [26,27,28,29,30]. The cryopreservation and regeneration process involves multiple stressors, including osmotic stress, chemical toxicity, ice crystallization, and cold shock, which collectively threaten cellular integrity and compromise preservation efficacy [24]. To mitigate these challenges, the three critical steps of preculture, cryoprotection, and optimized thawing form the foundation of successful cryopreservation. Preculture enables controlled cellular dehydration, reducing intracellular water content to enhance tolerance against rapid temperature fluctuations and extreme desiccation [27]. Cryoprotectants further improve preservation efficiency through synergistic mechanisms that establish a biological protection barrier during dehydration and low-temperature stress, primarily by inhibiting lethal ice crystal formation and maintaining membrane integrity [31]. Additionally, tailored thawing protocols prevent destructive ice recrystallization during rewarming, ensuring structural and functional recovery of preserved tissues [24]. Cryopreservation serves as a critical technique for the long-term and efficient conservation of forest tree germplasm resources. Its efficiency is co-regulated by multiple factors, including explant type, genotype, tissue culture procedure, and cryopreservation methodology [32]. Therefore, species-specific protocol optimization is essential to improve cryopreservation outcomes. Although L. olgensis is an important coniferous species in northern China, cryopreservation techniques for its EC have not yet been established. Our laboratory has successfully developed a stable SE system for L. olgensis [33], providing a consistent material basis for such studies. Therefore, the preservation of elite embryogenic cell lines is urgently required.
This study aims to establish a simple and efficient cryopreservation protocol by optimizing key steps in the cryopreservation process (including preculture, cryoprotectant, and thawing temperature). The goal is to maximize the retention of somatic embryogenic potential, thereby laying a crucial foundation for the long-term cryopreservation of elite germplasm resources in L. olgensis.
2. Results
2.1. Optimization of 2,3,5-Triphenyltetrazolium Chloride (TTC) Concentration for Cell Viability Assay
TTC is reduced by hydrogen in biological materials to form the insoluble red compound triphenylformazan. EC incubated in varying TTC concentrations under dark conditions for 24 h exhibited differential red coloration intensity (Figure 1A). The 0.5% TTC treatment yielded the deepest coloration, contrasting with the lightest color at 0.1%.
The TTC reduction assay is a widely used method for assessing cell viability [34]. The results showed highly significant differences in absorbance among the TTC concentrations tested (Figure 1B). The highest absorbance value was observed at 0.5% TTC (0.547), followed by 0.75% TTC (0.536), and the lowest at 0.1% TTC (0.182). These quantitative findings are consistent with the qualitative color observations.
2.2. Screening of Optimal Preculture and Cryoprotectant Protocols
2.2.1. Effects of Preculture and Cryoprotectant Protocols on Cell Viability
Range analysis (R-values, Table 1) revealed that the type of preculture medium had the greatest effect on the viability of L. olgensis EC (R = 1.931%), followed by preculture duration (R = 1.569%) and dimethyl sulfoxide (DMSO) concentration (R = 1.389%), while polyethylene glycol 6000 (PEG_6000_) concentration had minimal impact (R = 0.458%). Among all treatments, treatment 12 (a 24 h stepwise preculture on medium containing 0.2 and 0.4 mol∙L^−1^ sucrose, followed by cryoprotectant treatment with 0.4 mol∙L^−1^ sucrose, 5% (v/v) DMSO, and 10% PEG_6000_) resulted in significantly higher cell viability than the others.
Multiple comparisons (Table 2) showed that the highest viability (3.486%) was achieved with a 24 h preculture, which was significantly higher than the lowest viability (1.917%) observed at 12 h. No significant differences were found between 24 h and 36 h, or between 12 h and 48 h, but the difference between 24 h and 12 h was highly significant. Stepwise preculture on sucrose-containing medium resulted in a viability of 3.875%, which differed highly significantly from all other treatments. The highest viability under DMSO treatment (3.569%) was obtained at 2.5% concentration. This result was not significantly different from that of the 5% DMSO group, but was significantly lower than those treated with 10% and 15% DMSO. For PEG_6000_, the maximum viability (3.111%) occurred at 10% concentration, with no significant differences across the concentration gradient.
In summary, the optimal cryopreservation protocol for L. olgensis EC entails that the EC undergo 24 h preculture on a medium supplemented with 0.2 mol∙L^−1^ of sucrose, followed by a medium supplemented with 0.4 mol·L^−1^ sucrose. Subsequently, they are immersed in cryoprotectant solution containing 0.4 mol·L^−1^ sucrose, 2.5% DMSO, and 10% PEG_6000_. Since this specific combination was not included in the original 16 treatments of the orthogonal array testing strategy (OATS), further experimental validation was conducted to confirm its effectiveness in minimizing cryoinjury.
Under experimental conditions identical to the 16 OATS treatments, the same weight of EC (0.6 g) was cryopreserved using the optimized protocol. Cell viability was assessed with three replicates. Results demonstrated that EC preserved with the optimized protocol showed a post-thaw viability of 5.111% (Table 3). In contrast, the directly cryopreserved control showed only 1.111% viability, indicating a highly significant difference between the two groups.
2.2.2. Effects of Preculture and Cryoprotectant Protocols on Proliferation Rate
The proliferation rate of post-thaw EC reflects its recovery efficacy. This study quantified proliferation rates after 4 weeks of recovery for both the 16 OATS treatments and the optimized protocol (Table 4). The control exhibited no proliferation, whereas the optimized protocol achieved the highest proliferation rate (710%), followed by treatment 12 (480%). These results demonstrate that the optimized protocol minimizes cryoinjury and maximizes post-cryopreservation recovery.
2.3. Screening of Optimal Thawing Temperature
As shown in Figure 2, cell viability initially increased with rising thawing temperature, reaching a maximum of 5.11% at 37 °C, and then decreased at higher temperatures. The cell viability at 37 °C showed significant differences from that at all other temperatures tested. Therefore, 37 °C was selected as the optimal thawing temperature in this protocol.
2.4. Recovery Rate Assessment
Ten clumps of EC cryopreserved for 4 months using the optimized protocol (a 24 h stepwise preculture on medium containing 0.2 and 0.4 mol∙L^−1^ sucrose, followed by cryoprotectant treatment with 0.4 mol∙L^−1^ sucrose, 2.5% DMSO and 10% PEG_6000_) were thawed at 37 °C, and initial weights were recorded (Figure 3A,C). After recovery of 10 days, translucent white EC emerged. Following 4 weeks of recovery, all 10 clumps showed noticeable weight gain and were covered with a layer of translucent white callus (Figure 3B,D), confirming a 100% recovery rate. The emergence of new proliferative tissues confirms that the cryopreserved EC has successfully resumed normal growth under the optimized protocol.
2.5. Effect of Cryopreservation Duration on Cell Viability and Proliferation
To validate the long-term stability of this protocol for L. olgensis EC, samples cryopreserved for 1 day, 7 days, 1 month, 3 months, 6 months, and 1 year were thawed and assessed for cell viability (Figure 4A) and proliferation rate (Figure 4B). The results showed no significant differences in either cell viability or proliferation rate across the different cryopreservation durations, indicating negligible impact on post-thaw regenerative growth of EC. This protocol effectively minimized cryoinjury during extended cryopreservation, achieving stable long-term preservation of plant genetic resources.
2.6. SE and Germination After Cryopreservation
To evaluate the embryogenic potential after cryopreservation, EC that resumed normal proliferation was used in SE, germination, and plant regeneration assays. The cryopreserved EC retained the ability to undergo normal SE (Figure 5A). Crucially, somatic embryos derived from cryopreserved-recovered EC displayed normal morphology and developed into viable plantlets (Figure 5B–D). These results demonstrate that cryopreservation not only preserves proliferative competence but also maintains full embryogenic differentiation potential.
2.7. Flowchart of the Optimized Protocol
Based on the experimental results described above, a flowchart detailing the optimized cryopreservation procedure for L. olgensis EC is presented in Figure 6.
3. Discussion
This study successfully established a simple, efficient, and reliable protocol for the cryopreservation and regeneration of L. olgensis EC, addressing a critical gap in the preservation of this economically and ecologically important conifer.
A particularly notable result was the 100% recovery rate observed across all EC samples treated with the optimized protocol. Morphological evaluation showed complete encapsulation by translucent white callus after four weeks of recovery, highlighting the importance of integrating multiple protective strategies. The stepwise preculture using a sucrose gradient (0.2 mol∙L^−1^ followed by 0.4 mol∙L^−1^) under dark conditions at 25 °C promoted controlled cellular dehydration, which is essential for reducing osmotic shock and preventing intracellular ice formation during cryopreservation. This principle is supported by studies in other species, such as Castanea dentata [35], Castanea sativa [36], Robinia pseudoacacia [37], and Vitis spp. [38]. This was further enhanced by the composite cryoprotectant solution (0.4 mol∙L^−1^ sucrose, 2.5% DMSO, and 10% PEG_6000_). The combination of permeating (DMSO) and non-permeating (sucrose, PEG) cryoprotectants is consistent with established cryobiological theory [39]. Larix gmelinii var. [27] treated with this strategy (0.4 mol∙L^−1^ sorbitol + 5% DMSO) achieved a 70% regeneration rate. While the permeable agent DMSO is indispensable, its concentration must be carefully optimized to balance effectiveness and toxicity. In the present study, a lower concentration (2.5%) helped minimize DMSO cytotoxic effects, a concern also reported in Taxodium hybrid ‘zhongshanshan’ [40]. In the EC cell suspension of Taxodium hybrid ‘zhongshanshan’, DMSO concentrations were tested at 0%, 2.5%, 5%, 7.5%, 10%, and 12.5%. The results demonstrated that DMSO concentration significantly influenced both recovery time and callus proliferation. The most favorable recovery effect was observed with 10% DMSO following 60 min incubation on ice, which led to the shortest recovery period (5 days) and a high callus proliferation rate of 4.30. The inclusion of PEG is thought to enhance extracellular vitrification and stabilize membranes, further protecting cellular integrity [41]. Furthermore, rapid thawing in a 37 °C water bath resulted in the highest post-thaw viability (5.11%). This result is consistent with the fundamental cryobiological principle that rapid warming minimizes destructive ice recrystallization [42]. This aligns with findings in Olea europaea [43], and underscores the need for species-specific optimization at this step [12,27]. The highly significant difference in viability between the optimized protocol (5.11%) and direct cryopreservation controls (1.111%) clearly demonstrates the effectiveness of our protocol in reducing cryoinjury. While high TTC-reduction activity indicates viable cells with active metabolism, its discrepancy with proliferation capacity (e.g., in Treatment 1) highlights that only protocols that also preserve division competence, such as our optimized protocol, enable true long-term regeneration. A pivotal advantage of our protocol is its capability for long-term preservation without loss of biological quality. The absence of significant differences in cell viability and proliferation rate across cryopreservation durations from 1 day to 1 year confirms that cellular viability and proliferative capacity are stably maintained in liquid nitrogen. This stability is a cornerstone of successful cryopreservation [12]. It effectively eliminates the risks of somaclonal variation and loss of embryogenic competence inherent to chronic subculturing [6], thereby enabling the establishment of a reliable cryobank for L. olgensis genetic resources. Perhaps the most compelling evidence for the preservation of complete cellular function and totipotency is the ability of the regenerated EC to undergo normal SE. This ability confirms that the protocol not only preserves viability but also retains the differentiation capacity, which is essential for large-scale clonal propagation programs. The ability of the recovered EC to undergo SE is especially significant, given the previous work on L. olgensis that established methods for EC induction and SE [33], and Agrobacterium-mediated transformation [5].
In conclusion, this study develops the first comprehensive, highly efficient cryopreservation protocol for L. olgensis EC. Its success is evidenced by complete regeneration, stable long-term preservation, and maintained embryogenic potential. This protocol provides a powerful tool for the long-term conservation of elite genotypes of this important species. This supports advanced breeding programs, safeguards genetic diversity, and facilitates the deployment of clonal forestry. Future research could focus on employing techniques such as flow cytometry or SSR markers to conduct molecular verification of the genetic stability in L. olgensis following cryopreservation, and applying this validated protocol to a wider range of coniferous species.
4. Materials and Methods
4.1. Plant Material
The EC induced and subcultured for 6 months in our laboratory served as the experimental material [33,44]. The fresh EC selected and stabilized on proliferation medium for 3–5 days was used for subsequent experiments.
4.2. Culture Medium Formulation
All culture media (Table 5) were formulated using BM (Basal Medium) and MS (Murashige and Skoog) as the basal media [45].
4.3. Methods
4.3.1. Optimization of TTC Concentration
Cell viability refers to the percentage of living cells within a cell population, reflecting the overall health status of the cells [46]. Cell viability served as the key metric for determining the optimal cryopreservation protocol for EC [27].
Cell viability of L. olgensis EC was assessed using the TTC reduction assay, which primarily reflects dehydrogenase activity in living cells. The viability is estimated based on the intensity of red color formation resulting from the reduction of TTC to formation [47]. Fresh EC (0.1 g), not subjected to cryopreservation, was placed into 5 mL centrifuge tubes. Then, 1.5 mL of TTC solutions at concentrations of 0.1%, 0.25%, 0.5%, 0.75%, and 1% were added. The tubes were incubated in the dark at 25 °C for 24 h. Three independent replicates were performed for each concentration. After incubation, the staining intensity was visually assessed. The TTC solution was discarded, and any residual solution was removed by rinsing the EC with distilled water. EC was incubated in 3 mL of 95% ethanol at 65 °C for 30 min, and the absorbance of the resulting supernatant was then measured at 485 nm.
4.3.2. Preculture and Cryoprotectant Treatment
A total of 0.6 g EC was spread evenly onto a preculture medium for either stepwise preculture (first preculture on 0.2 mol∙L^−1^ sucrose or sorbitol, second preculture on 0.4 mol∙L^−1^ sucrose or sorbitol) or non-stepwise preculture (on 0.4 mol∙L^−1^ sucrose or sorbitol). All cultures were maintained in the dark at 25 °C for 12 h, 24 h, 36 h, or 48 h (Table 6). After preculture, the EC from different treatment groups were transferred into 2 mL cryovials, and 1 mL of filter-sterilized cryoprotectant solution was added at 25 °C. The cryoprotectant solution was formulated with DMSO (2.5%, 5%, 10%, 15%) and PEG_6000_ (0%, 5%, 10%, 15%) as base protectants, supplemented with 0.4 mol∙L^−1^ sucrose or sorbitol. The cryovials containing the cryoprotectant were then immediately transferred into a program cooling box (biosharp BS-02-CFC). The program cooling box commenced at a cooling rate of −1 °C∙min^−1^ until reaching −80 °C. After maintaining at −80 °C for at least 4 h, the cryovials were removed and rapidly plunged into a liquid nitrogen biological container for cryopreservation. Four factors, preculture duration, preculture strategy, DMSO concentration in the cryoprotective solution, and PEG_6000_ concentration, were evaluated, each at four levels. Sixteen treatment combinations were generated using OATS (Table 6). Cell viability (Section 4.3.1) and proliferation rate (Section 4.3.4) were assessed for each treatment after 24 h of cryopreservation. Following statistical analysis of viability across all treatment groups, the optimal preculture and cryoprotectant protocol was determined. A comparative analysis of proliferation rates across all treatment groups was performed, which validated the superior recovery performance of the optimal treatment group. EC without preculture and cryoprotectant treatment served as the directly cryopreserved control, with three replicates per treatment.
4.3.3. Thawing Treatment
After 24 h of cryopreservation, the cryovials were retrieved and thawed at 4 °C (refrigerator), 25 °C (ambient temperature), 37 °C, 42 °C, or 50 °C (water bath). Following complete EC thawing, the cryoprotectant solution was removed. The EC within each cryovial was washed 3 to 4 times with liquid proliferation medium. EC was then placed on sterile filter paper to absorb excess liquid before being transferred onto solid proliferation medium for subsequent cell viability assessment. Based on the cell viability (Section 4.3.1) results, the optimal thawing temperature was selected.
4.3.4. Proliferation Rate
The proliferation rate serves as a functional metric reflecting not only the survival of EC post-thaw but also the recovery capacity of the surviving cells. One day post-thawing, 0.1 g EC was randomly selected from the solid proliferation medium and transferred to fresh medium to thoroughly eliminate residual DMSO and PEG solutions. Following a 4-week recovery period, the samples were weighed under aseptic conditions using an analytical balance with a precision of 0.01 g to calculate the proliferation rate, with three replicates per treatment.
4.3.5. Effect of Cryopreservation Duration (Storage Duration in Liquid Nitrogen) on Cell Viability and Proliferation Rate
EC subjected to preculture and cryoprotectant treatment were preserved in liquid nitrogen for 1 day, 7 days, 3 months, 6 months, or 1 year. After each cryopreservation period, post-thaw cell viability (Section 4.3.1) and post-recovery proliferation rate (Section 4.3.4) were assessed to analyze the effect of duration on cryopreservation efficacy. Three independent replicates were established for each cryopreservation duration.
4.3.6. Assessment of Recovery Rate
The recovery rate serves as a reliable indicator of protocol stability. Ten cryovials cryopreserved for 4 months were retrieved and thawed. Under aseptic conditions, the L. olgensis EC from each cryovial was individually weighed using an analytical balance with a precision of 0.01 g. The EC was then transferred to solid proliferation medium and cultured in the dark at 25 °C. After 4 weeks, the EC was weighed again to assess recovery status. The recovery rate was calculated based on the number of recovery clumps.
4.3.7. Validation of Somatic Embryogenic Potential
Cryopreserved EC was thawed and subjected to proliferation according to the methods described in previous sections. Following at least one subculture cycle (15 days), 0.2 g EC was transferred onto pre-maturation medium. Cultures were maintained at 25 °C under dark conditions for 10 days. Subsequently, the EC was transferred to somatic embryo maturation medium. After approximately 40 days of culture on somatic embryo maturation medium, somatic embryos were carefully separated from the EC and transferred onto germination medium. Germination typically occurred within about 5 days. Following germination, root development was observed after approximately 20 days, leading to the regeneration of complete plantlets.
4.4. Statistical Analysis
The proliferation rate, cell viability, and recovery rate of cryopreserved EC were calculated using the following formulas:
The analysis of variance (ANOVA) was performed using SPSS Statistics 23.0. The OATS configuration was employed to identify the optimal combination of preculture and cryoprotectant conditions. The multiple comparisons were analyzed via the Least Significant Difference (LSD) program in SPSS Statistics 23.0. Values are presented as the mean ± SD.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1An P.Q. Cao Q. Wang C. Wang J.H. Zhang H.G. Zhang L. Spatiotemporal Expression and Bioinformatic Analyses of the HD-Zip Transcription Factor Family in Larix olgensis Plant Mol. Biol. Rep.20213921222510.1007/s 11105-020-01244-9 · doi ↗
- 2Zhao R.R. Qi S.Z. Cui Y. Gao Y. Jiang S.F. Zhao J. Zhang J.F. Kong L.S. Transcriptomic and physiological analysis identifies a gene network module highly associated with brassinosteroid regulation in hybrid sweetgum tissues differing in the capability of somatic embryogenesis Hortic. Res.20229 uhab 04710.1093/hr/uhab 04735031801 PMC 8788368 · doi ↗ · pubmed ↗
- 3Bai Y.H. Liu M. Zhou R. Jiang F.L. Li P. Li M.Q. Zhang M. Wei H.Y. Wu Z. Construction of ce RNA Networks at Different Stages of Somatic Embryogenesis in Garlic Int. J. Mol. Sci.202324531110.3390/ijms 2406531136982386 PMC 10049443 · doi ↗ · pubmed ↗
- 4Jiang S.F. Chen X.Y. Gao Y. Cui Y. Kong L.S. Zhao J. Zhang J.F. Plant Regeneration via Somatic Embryogenesis in Larix principis-rupprechtii Mayr Forests 202112133510.3390/f 12101335 · doi ↗
- 5Song Y. Bai X.M. Dong S.W. Yang Y.N. Dong H. Wang N.R. Zhang H.G. Li S.J. Stable and Efficient Agrobacterium-Mediated Genetic Transformation of Larch Using Embryogenic Callus Front. Plant Sci.20201158449210.3389/fpls.2020.58449233324434 PMC 7723890 · doi ↗ · pubmed ↗
- 6Caeiro A. Jarak I. Correia S. Canhoto J. Carvalho R. Primary Metabolite Screening Shows Significant Differences between Embryogenic and Non-Embryogenic Callus of Tamarillo (Solanum betaceum Cav.)Plants 202312286910.3390/plants 1215286937571022 PMC 10420837 · doi ↗ · pubmed ↗
- 7Vásquez-Rivera A. Sommer K.K. Oldenhof H. Higgins A.Z. Brockbank K.G.M. Hilfiker A. Wolkers W.F. Simultaneous monitoring of different vitrification solution components permeating into tissues Analyst 201814342042810.1039/C 7AN 01576 C 29236110 · doi ↗ · pubmed ↗
- 8Hu J.W. Pu Z.Y. Hao C.H. Yan H.L. Qian S.M. Zhu T.Q. Ma W.J. An S.P. Kong L.S. Wang J.H. Optimized cryopreservation of embryogenic tissue of Picea abies based on differential scanning calorimetry Cryobiology 202512010527810.1016/j.cryobiol.2025.10527840614588 · doi ↗ · pubmed ↗