A Second Opportunity for the Peptide-Based Analogues with γ-Lactam at the P1 Position: Human Cathepsin S Inhibition
Santo Previti, Nunzio Iraci, Elsa Calcaterra, Roberta Ettari, Maria Zappalà

TL;DR
Researchers found that some SARS-CoV-2 inhibitors also block human cathepsin S, suggesting new drug possibilities.
Contribution
Discovery that γ-lactam-based SARS-CoV-2 inhibitors can inhibit human cathepsin S, offering a new drug design strategy.
Findings
Peptide-based analogues with γ-lactam at P1 inhibit human cathepsin S.
Molecular simulations suggest γ-lactam can form water-mediated hydrogen bonds to enhance inhibition.
Repurposing these inhibitors could lead to potent and selective hCatS ligands.
Abstract
Background/Objectives: SARS-CoV-2 pandemic led to the identification of peptide-based main protease (Mpro) inhibitors. The overwhelming majority of them carry an electrophilic warhead and a γ-lactam at the P1 position. During the selectivity assessment of an in-house Michael acceptors targeting SARS-CoV-2 Mpro, we unexpectedly observed a significant inhibition of human cathepsin S (hCatS). Methods: The biological investigation of three compounds (i.e., SPR38, SPR39, and SPR41) against hCatS was performed. The binding mode of SPRs was investigated by docking and molecular dynamics simulations. Results: Biological investigation has corroborated that hCatS is sensitive to peptide-based analogues harbouring γ-lactam at the P1 position and a vinyl methyl ketone warhead. In silico studies revealed that despite being solvent exposed, the γ-lactam at P1 might be involved in water-mediated…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
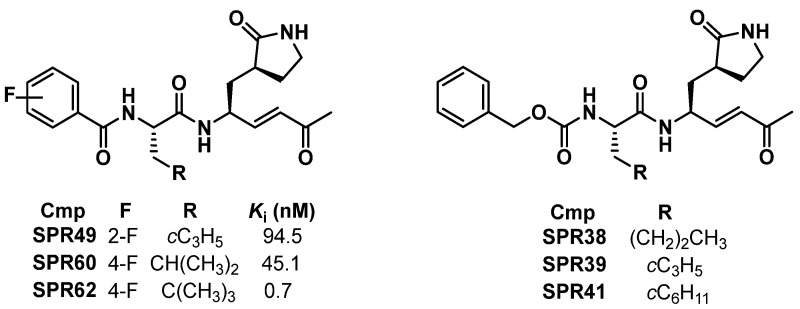 Figure 1
Figure 1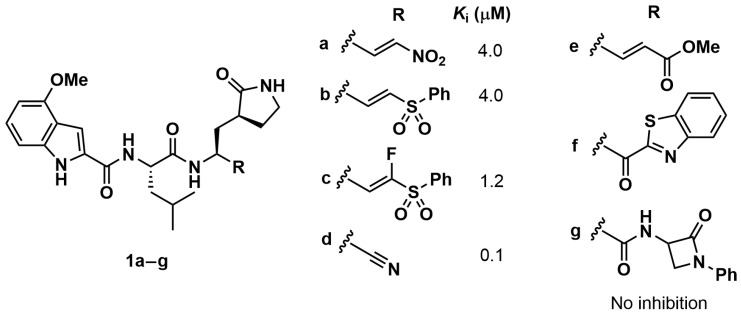 Figure 2
Figure 2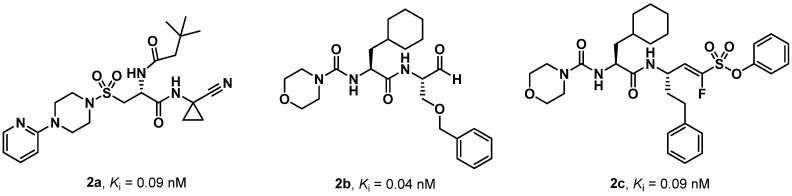 Figure 3
Figure 3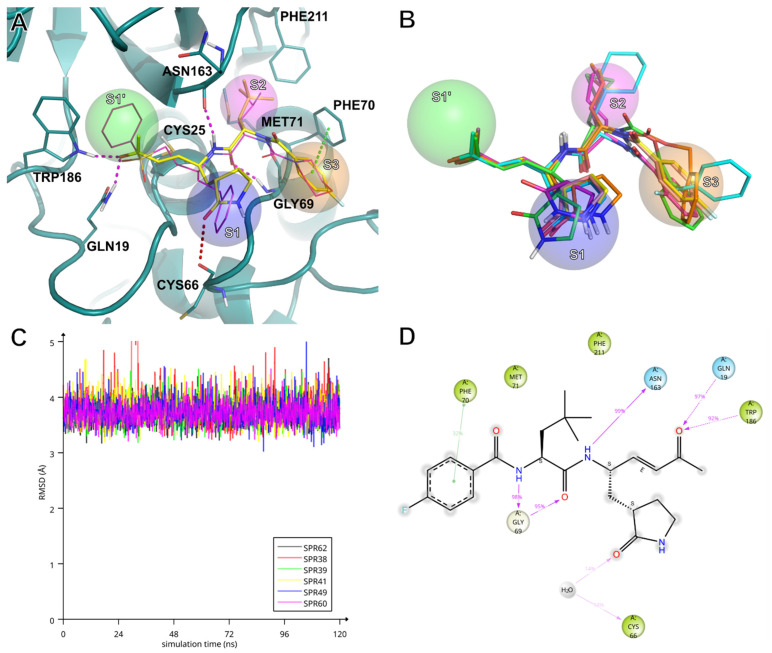 Figure 4
Figure 4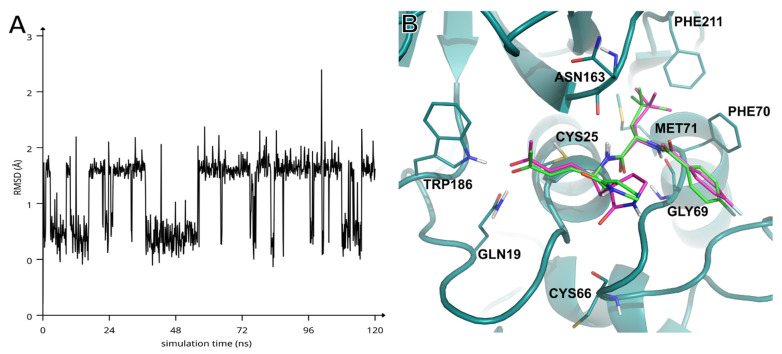 Figure 5
Figure 5 Figure 6
Figure 6- —Italian Ministry of University and Research
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBiochemical and Structural Characterization · RNA and protein synthesis mechanisms · Antimicrobial Peptides and Activities
1. Introduction
Papain-like proteases are lysosomal proteolytic enzymes that have been identified in a variety of organisms, including animals, invertebrates, plants, and microorganisms [1]. In mammals, these types of proteases are commonly known as cathepsins. The human genome encodes for fifteen cathepsins, which can be classified as serine, aspartic, or cysteine proteases according to the amino acid responsible for the nucleophilic attack [2]. Cathepsins mediate a variety of physiological processes, including extracellular matrix degradation, intracellular protein turnover, neuronal cell homeostasis, and the processing of damaged proteins, immunoglobulins, and prohormones [3,4,5].
Human cathepsin S (hCatS or CTSS) is a cysteine protease biosynthesized as a pre-proenzyme of 331 amino acid residues [6]. The mature enzyme is constituted of a single-chain polypeptide comprising 217 amino acids and featuring the catalytic triad Cys25/His164/Asn184 [7]. hCatS is involved in multiple physiological and pathological processes, including inflammation [8], metabolic and neurological disorders [9], cancer [10], cardiovascular [11], renal [12], respiratory [13], and autoimmune diseases [14]. In addition to the lysosomal compartments, hCatS has also been detected extracellularly, where it has been shown to promote the degradation of extracellular proteins and angiogenesis [15]. Finally, this cysteine protease plays a pivotal role in regulating antigen processing and presentation through the major histocompatibility complex (MHC) II pathway in antigen-presenting cells (APCs) [16,17,18].
Given its involvement in various diseases, hCatS has emerged over the past decades as a promising therapeutic target and biomarker [19,20]. Several Structure–Activity Relationship (SAR) studies have resulted in the identification of interesting hCatS inhibitors [21,22,23,24,25], typically identified starting from compounds with potent inhibitory properties towards other human cysteine proteases and subsequently optimized through lead refinement strategies.
Recently, we reported the identification of dual SARS-CoV-2 M^pro^/human cathepsin L (hCatL) inhibitors [26]. Selectivity assays over a panel of human cysteine proteases revealed Ki values in the nanomolar and sub-nanomolar range towards hCatS for SPR49, SPR60, and SPR62, as illustrated in Chart 1. The three Michael acceptors carry a methyl vinyl ketone warhead, a γ-lactam at the P1, aliphatic amino acids at the P2 position, and a mono-fluorinated benzoyl ring as the N-cap. It is well established that the presence of Gln and its cyclic analogue (3S)-pyrrolid-2-one-3-yl-L-alanine adjacent to the cleavage site of SARS-CoV-2 M^pro^ was found to be essential for effective inhibition [27,28]. Since no known human proteases have the same cleavage specificity as SARS-CoV-2 M^pro^ [29], the such potent inhibition observed against hCatS was unexpected.
In light of these findings, we herein report the biological investigation of three in-house (3S)-pyrrolid-2-one-3-yl-L-alanine-containing Michael acceptors (i.e., SPR38, SPR39, and SPR41, shown in Chart 1) [30] towards hCatS, along with a discussion of SAR and molecular docking studies of the most promising inhibitors.
2. Results and Discussion
2.1. Biology
SPR38, SPR39, and SPR41 were assayed against hCatS using Cbz-VVR-AMC as the fluorogenic substrate. All analogues exhibited potent inhibitory properties towards the target, with Ki values in the double-digit nanomolar range (Table 1). SPR38 and SPR39, which feature norleucine (Nle) and cyclopropylalanine (Cpa), respectively, at the P2 site, showed comparable binding affinity. In contrast, the cyclohexylalanine (Cha)-derivative SPR41 exhibited the most potent inhibitory effect (Ki = 10.7 ± 1.1 nM). The presence of Cha in SPR41 resulted in 4- and 5-fold improvement in Ki values compared to Nle and Cpa in SPR38 and SPR39, respectively. Compared to our previously reported SPR analogues targeting hCatS [26], only SPR41, with Cha at the P2 site, showed a Ki value in the low nanomolar range, similar to those observed for SPR60 and SPR62.
The inhibitory properties of SPRs against hCatS were determined using Ki values, consistent with previously reported literature data [26,30]. Although both dialysis and dilution assays confirmed a covalent and irreversible binding mode of SPRs against SARS-CoV-2 M^pro^, hCatL, hCatB, and hCatS, time-dependent inhibition profiles did not exhibit sufficient curvature to allow accurate determination of the kinetic parameters Ki, kinact, and k2nd. These findings support a bi-phasic inhibition mechanism, whereby high inhibitor concentrations promote irreversible inactivation of the enzyme, whereas at lower concentrations, the interaction remains predominantly reversible.
The three analogues showed notable selectivity towards hCatS. In fact, inhibitory properties in the micromolar range towards hCatL and hCatB were observed, except for SPR41, for which submicromolar activity against hCatL was determined. This trend highlights the contribution of the common binding region of the SPRs, namely the methyl vinyl ketone warhead, a γ-lactam residue and an aliphatic amino acid at the P1 and P2, respectively, provided for strong hCatS inhibition with Ki values in the low nanomolar or even subnanomolar range. Conversely, hCatL and hCatB appear to be less sensitive to SPRs. Except for the Leu-containing derivative SPR60, the other Michael acceptors displayed only moderate activity towards hCatL, with Ki values in the submicromolar and micromolar range. For hCatB, even higher values of Ki were observed, resulting in selectivity indices ranging from 157 to 11 000. These findings indicate that SPRs are highly compatible with the catalytic site of hCatS, while exhibiting markedly reduced affinity for hCatL and, in particular, hCatB.
To the best of our knowledge, no hCatS inhibitors incorporating (3S)-pyrrolid-2-one-3-yl-L-alanine have been rationally developed to date. However, Schirmeister and co-workers assayed a series of peptide-based compounds bearing a Gln pentatomic surrogate at the P1 position towards hCatS (Chart 2) [32]. Analogues 1a–d showed Ki values in the micromolar and submicromolar range, whereas derivatives 1e–g showed no measurable inhibitory activity. Since these compounds differ exclusively in the warhead, it is evident that this moiety plays a crucial role for hCatS inhibition. Specifically, the nitro and sulfonyl phenyl moieties of Michael acceptors 1a and 1b–c, respectively, as well as the nitrile warhead of 1d, were found to be suitable for hCatS inhibition, whereas, the presence of vinyl methyl ester (1e), benzothiazole (1f), and β-lactam ring (1g) warheads were ineffective. Based on these findings reported in the literature and our results with SPR analogues, the methyl vinyl ketone warhead appears highly suitable for hCatS inhibition, regardless of surrounding binding regions. Notably, Leu derivative SPR60 showed a single-digit Ki value in the nanomolar range, representing a two/three-orders-of-magnitude improvement over Leu-analogues 1a–d. Moreover, the presence of the further branched amino acid (i.e., tert-butylalanine, known as Tba, in SPR62) enhanced hCatS affinity and conferred remarkable selectivity over hCatL (SI = 1000) and hCatB (SI = 11 000).
As previously mentioned, several potent hCatS inhibitors have been rationally developed. Here, we compare some of these reference compounds with the most active SPR analogues, namely SPR41, SPR60, and SPR62. The vinyl sulfone K11017 (Mu-Leu-hPh-VSPh), also known as LHVS, is widely employed as a positive control in hCatS inhibition assays [31]. SPR41 and SPR60 showed binding affinities comparable to K11017, whereas SPR62 exhibited a Ki value approximately one order of magnitude lower. A similar trend was observed when comparing SPRs with BI-1124 and BI-1195, two peptidomimetics carrying the nitrile warhead developed by Boehringer Ingelheim. Both compounds are used as reference inhibitors of hCatS, with a Kd value of 9 and 31 nM. Notably, SPR62, the most potent and selective SPRs (Ki = 0.7 nM), exhibited a Ki value only one order of magnitude higher compared to compounds 2a–c (Chart 3) [22,23,33], which represent some of the most promising hCatS inhibitors rationally developed in three different SAR studies and known in the literature.
During the SARS-CoV-2 pandemic, drug repurposing emerged as a key strategy for the rapid development of effective antiviral agents [34,35]. The SPR analogues discussed herein was originally developed as potential SARS-CoV-2 M^pro^ inhibitors, with their hCatS inhibitory properties identified only during selectivity profiling. This observation suggests that further promising hCatS inhibitors could be discovered among compounds initially developed for SARS-CoV-2 M^pro^. Indeed, an impressive number of SAR studies have reported the development of peptide-based SARS-CoV-2 M^pro^ inhibitors carrying an electrophilic warhead, the (3S)-pyrrolid-2-one-3-yl-L-alanine residue at the P1 site, an aliphatic amino acid at the P2 position, and with further optimization targeting the S3, S4, and S5 subsites [36]. The biological investigation of these molecules towards hCatS could provide valuable insights and enable the identification of novel building blocks and warheads useful for the hCatS inhibition. In fact, a wide range of warheads have been successfully investigated for SARS-CoV-2 M^pro^ inhibition, including vinyl sulfonamide, aldehyde, nitrile, Michael acceptors, ketoamide, 4-chlorobut-2-ynamide, hydroxymethylketone, and benzyloxymethylketone, amongst others [37,38,39]. While nitriles, Michael acceptors, and aldehydes, have been studied for hCatS inhibition, the potential of diverse reactive warheads remains largely unexplored. Furthermore, the nature of the electron-withdrawing groups (EWGs) in Michael acceptors significantly influences hCatS inhibitory potency, as evidenced by comparison among SPR62, K11017, 1a–c, 1e, and 2a. Before the SARS-CoV-2 pandemic, the incorporation of γ-lactam at the P1 site of peptidomimetics was restricted to the development of antiviral agents. Considered the promising results obtained with SPRs, (3S)-pyrrolid-2-one-3-yl-L-alanine-containing compounds could be rationally developed for hCatS inhibition. Drug/molecule repurposing of compounds originally designed as SARS-CoV-2 M^pro^ inhibitors could therefore accelerate the identification of promising warheads, building blocks, and lead compounds for hCatS inhibition.
2.2. Docking Studies
Compounds SPR38, SPR39, SPR41, SPR49, SPR60, and SPR62 were investigated in silico for their molecular recognition at the active site of hCatS using the extended sampling protocol of the Schrödinger Induced Fit Docking (IFD) tool [40]. As expected, docking results confirmed that the P1, P2, and P3 residues appropriately occupy the S1, S2 and S3 sub-pockets of hCatS (Figure 1A,B—IFD poses can be found in Supplementary Materials, in PDB format). It was observed that the β-carbon atoms of the inhibitors’ warhead stably positioned in close proximity to the γ-sulphur of the catalytic Cys25 throughout 120 ns-long molecular dynamics (MD) simulations (Figure 1C). Overall, all SPRs showed highly similar interaction patterns throughout the MD simulations (Figure S1). Several studies have addressed which face is involved in the nucleophilic attack and, thereby, the configuration of the new chirality centre at the sp3-hybridized carbon [41,42,43]. In our case, it was predicted that the nucleophilic attack of the Cys25 thiolate occurs from the re face of the inhibitors. Consequently, the pivotal reaction between the Cys25 thiolate and prochiral electrophilic β-carbon would result in the formation of an (S)-chirality centre. Figure 1C depicts the key interactions between hCatS and SPR62 during the MD simulation, highlighting stable contacts with multiple residues within the active site. At the P2 position, Tba residue of SPR62 forms strong hydrogen bonds with the backbone of Gly69 through both its NH and carbonyl oxygen. The P1 NH group of SPR62 acts as a hydrogen bond donor, interacting with the backbone carbonyl of Asn163. The warhead’s carbonyl oxygen accepts two additional hydrogen bonds from the side chain NH groups of Gln19 and Trp186. Futhrer stabilizing interactions include π–π stacking with Phe70 and hydrophobic contacts with Met71 and Phe211. MD trajectory analysis revealed that the γ-lactam moiety, although solvent-exposed and lacking direct residue contacts, occasionally engages in a water-mediated hydrogen bond between its carbonyl oxygen and that of Cys66. At the same time, ligand’s root mean square deviation (RMSD) analysis indicates that SPR62 is plausibly oscillating between two bound conformations over the course of the simulation (Figure 2A). Notably, the γ-lactam carbonyl oxygen exhibits the highest root mean square fluctuation (RMSF) value among all atoms of SPR62 (1.863 Å), supporting the hypothesis that the conformational variability arises mainly from alternative orientations of the γ-lactam ring.
To further explore this behaviour, we performed cluster analysis on the trajectory (see Supplementary Materials), which identified two significantly populated conformational clusters (Table S1). The clusters contained SPR62 mainly differing for the orientation of the γ-lactam carbonyl group: either pointing “up” (Figure 2B, yellow sticks, cluster 1) or “down” toward the carbonyl of Cys66 (Figure 2B, magenta sticks, cluster 2). Single-point energy calculations (in vacuum) averaged over all ligand conformations within each cluster revealed that the “down” conformation carries an energetic penalty of approximately 3 kcal/mol (Table S1). However, this cost may be rewarded by water-mediated hydrogen bonds involving Cys66 or neighbours such as Lys64 or Asn67 (Figure S1).
Despite this energy difference, both the two conformational clusters are significantly populated (539 vs. 409), suggesting a dynamic equilibrium. This observation warrants future investigation into the role of structured water molecules near Cys66 and their potential exploitation to enhance potency and selectivity toward hCatS.
3. Materials and Methods
3.1. Biological
The biological assays against hCatS were performed in accordance with the methods previously reported by Prof. Schirmeister’s research group [23,33]. Briefly, the inhibitory activities of SPR38, SPR39, and SPR41 against hCatS (purchased form Merck, Darmstadt, Germany) were evaluated using the fluorogenic substrate Cbz-Val-Val-Arg-AMC (Bachem, Bubendorf, Switzerland). The fluorescence was recorded in white flat-bottom 96-well plates (Greiner Bio-One, Kremsmünster, Austria) with a Tecan Spark 10M plate reader (Tecan Group Ltd., Männedorf, Switzerland). Stock solutions of the substrate and SPRs were prepared in DMSO. Each well contained 200 µL, consisting of 180 µL assay buffer, 5 µL enzyme solution in buffer, 10 µL inhibitor in DMSO (or DMSO alone as negative control), and 5 µL substrate. Assay buffer consisted of 50 mM potassium phosphate, 2.5 mM DTT, and 2.5 mM EDTA at pH 6.5, while the enzyme buffer consisted of 35 mM potassium phosphate, 35 mM sodium acetate, 2 mM DTT, and 2 mM EDTA at pH 6.5. All chemicals and reagents used for enzyme and assays buffer, along with DMSO, were purchased from Merck (Darmstadt, Germany). The enzyme (10 nM) was diluted and pre-incubated in enzyme buffer at room temperature for 20 min before trnsfer to the assay buffer. The reaction was initiated by adding the substrate (10 µM) under rigorous mixing. The reaction was monitored for 10 min at 25 °C, and fluorescence readout was performed in 30 sec intervals. AMC release was detected using an excitation wavelength of 380 nm and an emission wavelength of 460 nm. Residual enzymatic activity was calculated from the substrate hydrolysis rate in the presence of inhibitors relative to the DMSO control. To determine the IC_50_ values dilution series of the inhibitors were prepared (eg. final concentrations: 100, 30, 10, 3, 1, 0.3, 0.1 μM, and DMSO as control). The IC_50_ values were calculated using GRAFIT [44] by fitting the residual enzymatic activity to the four-parameter IC_50_ equation (Equation (1)):
where Y is the substrate hydrolysis rate, Y_max_ is the maximum value of the dose response curve at inhibitor concentrations [I] = 0 µM, Y_min_ is the minimum value at high inhibitor concentrations, and s is the Hill coefficient [45].
To calculate the Ki value, Equation (2) was used:
with Km as the substrate concentration at which the hydrolysis activity is half maximal and [S] as the substrate concentration [45].
3.2. Molecular Modelling
3.2.1. Target Structure Preparation
The experimental structure of hCatS in complex with K11017 (PDB ID: 1NPZ [41]) was obtained from the RCSB Protein Data bank [46]. The covalently bound inhibitor was initially removed, and the structure was processed using the Schrodinger Protein Preparation workflow to optimize the target structure for the following studies [47]. Following previous protocols [48], hydrogen atoms were added and bond orders were assigned. The hydrogen-bonding network was optimized by reorienting hydroxyl and thiol groups, the amide groups of Asn and Gln, and imidazole rings of His residues. The ionization and tautomeric states of His, Asp, Glu, Arg, and Lys residues were adjusted to match physiological pH of 7.4 using PROPKA. The structure was then submitted to a minimization (OPLS2005 force field [49]) that was stopped when RMSD of heavy atoms reached 0.30 Å. All crystallographic water molecules were removed prior to further analysis.
3.2.2. Preparation of Ligands
Ligands were sketched using the Maestro GUI v2025-2 [50] and were then submitted to a conformational search using MacroModel v2025-2 [51]. A Low-Mode Conformational Search (LMCS) method was performed, with a maximum of 1000 steps per structure. Calculations were carried out in implicit solvent (GB/SA) and energy minimization was performed on each conformer using the PRCG method, with a maximum of 2500 iterations per step. For each ligand, only the global minimum energy conformation was retained for subsequent docking simulations.
3.2.3. Molecular Docking Simulations
The Schrödinger Induced Fit Docking (IFD) v2025-2 workflow was used to explore the binding mode of the ligands [40]. Residues within 5 Å of the experimentally bound conformation of K11017 were selected as flexible. In the initial docking stage, the docking space was centred on the crystallographic pose of K11017, and defined as a (30 Å)^3^ cubic box, with a smaller, nested, (15 Å)^3^ cubic one where the diameter midpoint of each docked ligand was required to stay. After the Prime refinement of flexible residues [52], the second docking stage (redocking) was performed assigning the outerbox size automatically, based on the docking pose from the first docking stage. Both first docking and redocking were performed using GlideSP v2025-2 [53]. Final docking poses were scored and ranked using the default IFDscore, the best scoring complex for each ligand was used as input for the molecular dynamics simulations.
3.2.4. Molecular Dynamics Simulations
MD simulations were set and run using Desmond v2025-2 [54]. Simulated environments were built using the TIP3P water model [55] and the OPLS 2005 force field [49]. Prior to the 120 ns-long production stage, the systems were relaxed using the default multistep protocol. After system relaxation, 120 ns-long MD simulations were carried out at a 300 K in the NPT ensemble using a Nose-Hoover chain thermostat and a Martyna-Tobias-Klein barostat (1.01325 bar), applying a 1 kcal/mol harmonic constrain on the backbone heavy atoms. Time steps were set to 2 fs, 2 fs and 6 fs for bonded, near, and far interactions, respectively. Structures were sampled every 120 ps and trajectories were analyzed using Maestro [50] and VMD [56].
3.2.5. Trajectory Clustering and Energy Evaluation of Conformers
Snapshots of the SPR62/hCatS complex conformations were extracted from the MD trajectory. Conformations were superimposed by the backbone heavy atoms of hCatS and then SPR62 bound conformations were clustered using the conformer_cluster.py script [57], based on atomic RMSD values. RMSD calculations were performed in place (using the previous hCatS backbone alignment) on the amide motif (excluding the hydrogen atom) of the γ-lactam. Merge distance cutoff was set to 2 Å, using the average linkage clustering method. SPR62 conformations were evaluated for their ligand energies (in vacuum), and the energies of each conformation were averaged for each of the two most populated clusters.
4. Conclusions
Molecules originally developed as SARS-CoV-2 M^pro^ inhibitors showed potent affinity towards hCatS. These compounds carry a methyl vinyl ketone warhead, a γ-lactam Gln derivative at the P1 site, different aliphatic amino acids at the P2 site, and either a fluorine-containing phenyl ring or Cbz as the N-capping. Herein, SPR38, SPR39, and SPR41, differing only in their P2 residue, showed a relevant double-digit nanomolar affinity for hCatS, along with selectivity indices ranging from 24 to 74 over hCatL and from 175 to 1354 and hCatB. The biological data herein reported confirm the significant inhibitory properties against hCatS of peptide-based Michael acceptors originally developed for SARS-CoV-2 M^pro^. All the SPR derivatives share the P1-warhead fragment, namely the (3S)-pyrrolid-2-one-3-yl-L-alanine at the P1 site and a methyl vinyl ketone warhead. Comparison with known hCatS inhibitors highlighted this fragment as a key determinant for hCatS inhibition. Molecular modelling studies provided insight into the role of the solvent-exposed P1 γ-lactam substituent in target recognition. Furthermore, in silico analyses also suggested two additional strategies for the potential optimization of potency and selectivity toward hCatS, such as i) the design of modified P1 substituents capable to make more efficient water mediated H-bonds, or ii) the introduction of extended substituents able to displace structural water molecules and establish direct interactions with residues such as Cys66. Overall, peptide-based Michael acceptors, originally developed for SARS-CoV-2 M^pro^, demonstrated potent and selective inhibition of hCatS. Since a large number of these compounds carries a γ-lactam Gln derivative at the P1 and an electrophilic warhead, the repurposing strategy of molecules with this fragment could provide significant data for hCatS inhibition.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bromme D. Papain-like cysteine proteases Curr. Protoc. Protein Sci.20012121.2.121.2.1410.1002/0471140864.ps 2102 s 2118429163 · doi ↗ · pubmed ↗
- 2Patel S. Homaei A. El-Seedi H.R. Akhtar N. Cathepsins: Proteases that are vital for survival but can also be fatal Biomed. Pharmacother.201810552653210.1016/j.biopha.2018.05.14829885636 PMC 7172164 · doi ↗ · pubmed ↗
- 3Pecar Fonovic U. Kos J. Mitrovic A. Compensational role between cathepsins Biochimie 2024226627610.1016/j.biochi.2024.04.01038663456 · doi ↗ · pubmed ↗
- 4Turk V. Stoka V. Vasiljeva O. Renko M. Sun T. Turk B. Turk D. Cysteine cathepsins: From structure, function and regulation to new frontiers Biochim. Biophys. Acta 20121824688810.1016/j.bbapap.2011.10.00222024571 PMC 7105208 · doi ↗ · pubmed ↗
- 5Yadati T. Houben T. Bitorina A. Shiri-Sverdlov R. The ins and outs of cathepsins: Physiological function and role in disease management Cells 20209167910.3390/cells 907167932668602 PMC 7407943 · doi ↗ · pubmed ↗
- 6Lecaille F. Kaleta J. Bromme D. Human and parasitic papain-like cysteine proteases: Their role in physiology and pathology and recent developments in inhibitor design Chem. Rev.20021024459448810.1021/cr 010165612475197 · doi ↗ · pubmed ↗
- 7Geetha D. Hameeda B.A. Jose D. Kuriakose N. Skaria T. Novel insights into the dynamic conformational transitions and active site plasticity of human immunoregulatory cathepsin S Proteins 2025931805181810.1002/prot.2684540418096 · doi ↗ · pubmed ↗
- 8Vizovišek M. Vidak E. Javoršek U. Mikhaylov G. BratovšA. Turk B. Cysteine cathepsins as therapeutic targets in inflammatory diseases Expert Opin. Ther. Targets 20202457358810.1080/14728222.2020.174676532228244 · doi ↗ · pubmed ↗