Molecular Typing of Acanthamoeba Using Mitochondrial rDNA Spacers
Daniele Corsaro

TL;DR
This study explores the use of mitochondrial rDNA spacers for molecular typing of Acanthamoeba, a parasitic amoeba, to better distinguish between closely related species.
Contribution
The study introduces mitochondrial rDNA spacers as a novel and effective marker for molecular typing of Acanthamoeba species.
Findings
Lineage-specific profiles were identified for MG2 and MG3 species using mitochondrial spacers.
MG1 species have a distinct tRNA gene arrangement compared to other groups.
Phylogenetic analysis using mitochondrial spacers aligns with rDNA-based classification.
Abstract
Acanthamoeba is a widespread free-living amoeba known as an opportunistic parasite of humans and other animals. It comprises several species, whose characterisation relies currently on the analysis of 18S rDNA sequences, recognising more than twenty genotypes; however, the distinction between closely related lineages remains unclear. In this study, the spacer region between the mitochondrial large and small subunits of rRNA genes was analysed for its usefulness as a marker for molecular typing. Previous studies have shown that the mitochondrial spacer contains a group of five transfer RNA (tRNA) genes, and that its length and sequence vary considerably between strains. A total of forty-two mitochondrial spacers were examined here, including twenty-five newly recovered sequences, from ten genotypes covering the three morphological groups of Acanthamoeba. The results showed that…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
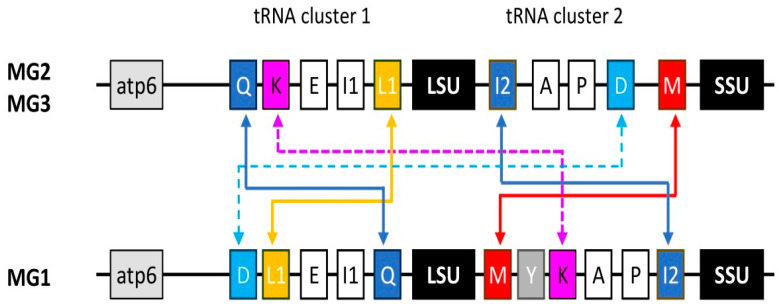 Figure 1
Figure 1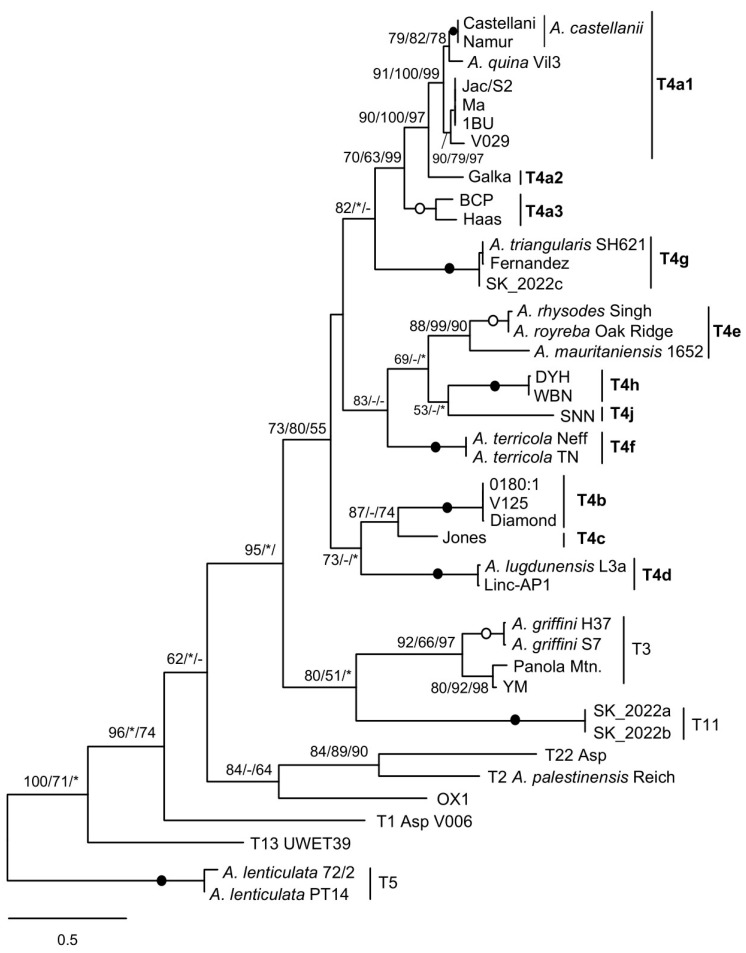 Figure 2
Figure 2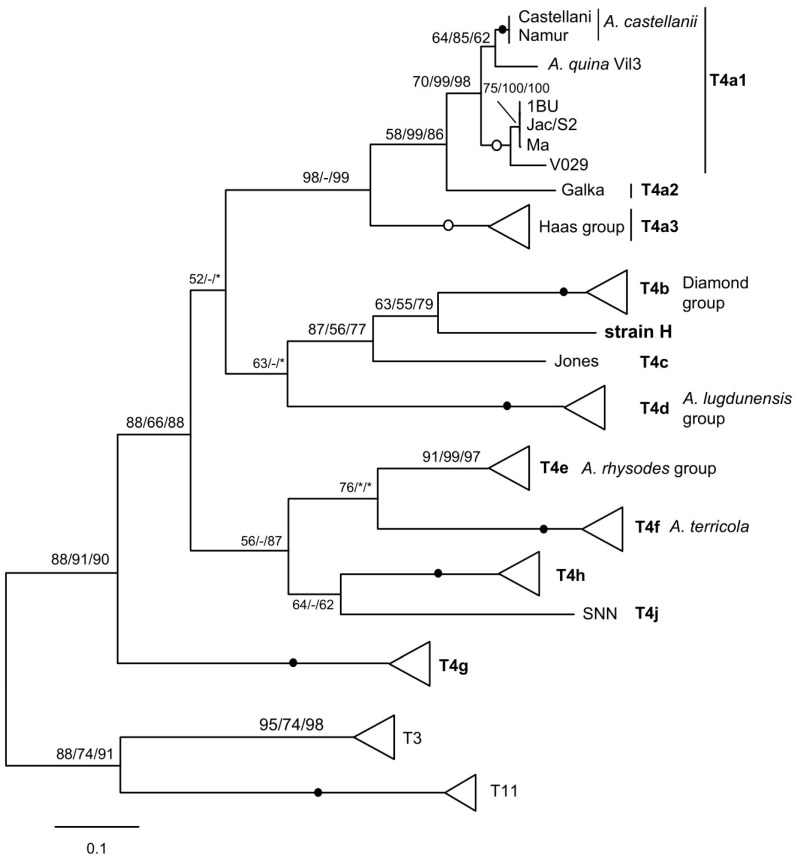 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLegionella and Acanthamoeba research · Yersinia bacterium, plague, ectoparasites research · Protist diversity and phylogeny
1. Introduction
Acanthamoeba (Amoebozoa, Discosea, Centramoebida, Acanthamoebidae) is a genus of free-living amoebae, distributed worldwide in both aquatic and terrestrial environments. Its life cycle includes an active trophozoite feeding on bacteria and other microbes, and a highly resistant, double-walled dormant cyst. It deserves special attention because it can act as an opportunistic parasite in humans and other vertebrates, primarily causing amoebic keratitis (AK), leading to vision loss, and fatal granulomatous amoebic encephalitis (GAE), often due to the hematogenous spread of a pulmonary or skin infection [1,2]. Disease severity can vary considerably depending on the patient’s health status and the pathogenicity of the amoeba strain. Indeed, Acanthamoeba exhibits high genetic and pathogenic variability, with some species appearing more virulent than others, although a clear relationship has not been established. Furthermore, Acanthamoeba frequently harbours endosymbionts, including pathogens such as Legionella, thereby contributing to their dissemination [3].
Acanthamoeba comprises several species, divided into three morphological groups (MG1 to MG3) based on cyst characteristics. MG1 species have a stellate endocyst with a well-separated ectocyst, while MG2 species may have a stellate, polygonal, or nearly spherical endocyst with a usually wrinkled ectocyst, and MG3 species have a round endocyst with a usually smooth ectocyst [4]. Acanthamoeba lineages are currently reorganised into more than twenty genotypes (T1 to T23) by molecular phylogenetic analysis based on the nuclear small subunit (SSU) ribosomal RNA gene (18S rDNA). The molecular approach allows the recovery of both classical and novel species, also revealing some inconsistencies based on previous morphological identifications, which have led to numerous strain misassignments [5,6]. Analysis of 18S and large subunit (LSU) nuclear rDNA sequences indicates greater diversification of Acanthamoeba, with more undescribed lineages that could represent new genotypes or even new genus-level taxa [7]. However, the vast majority of strains isolated from clinical and environmental samples belong to the T4 genotype, a large group comprising closely related strains and including the type strains of several MG2 species. Improving the resolution of phylogenetic relationships within closely related lineages is clearly useful for taxonomic purposes, but it is also useful for more reliable diagnosis and epidemiological investigation. In the case of T4, various nuclear and mitochondrial SSU rDNA subtypes have been defined, with good consistency between them [6,8]. They are based on complete sequences, approximately 2300–2600 or 1500 bp for the nuclear or mitochondrial SSU, respectively, and currently comprise eight nuclear subtypes (T4A to T4H) and ten mitochondrial subtypes (T4a to T4j) [6,8,9,10]. Promising results have also been obtained using the rapidly evolving internal transcribed spacer (ITS) region of the nuclear rDNA operon [11,12,13]. In almost all eukaryotes, this region contains the 5.8S rRNA gene, which is separated from the SSU and LSU rRNA genes by ITS1 and ITS2, respectively. It is used as a whole to distinguish, for example, species and genotypes of Naegleria (Discoba, Heterolobosea), a potentially pathogenic amoeboflagellate [14], whereas variations in the eukaryotic core secondary structure of ITS2 correlate with lineage separation [15].
Another potentially useful marker for differentiating Acanthamoeba strains could be the spacer between the mitochondrial LSU and SSU rRNA genes, which closely resembles the bacterial spacer except that the rRNA genes are inverted. In prokaryotes, the ITS often includes transfer RNA (tRNA) genes, while the homologous part of 5.8S is included in the 5’ end of the LSU [16,17], and the ITS region is increasingly used for the identification of bacteria [18,19]. In the Neff strain, now Acanthamoeba terricola [20], this region contains five tRNA genes [21]. By analysing the mitochondrial ITS of sixteen other strains belonging to four genotypes (T1, T2, T3, T4), Ledee and Byers [22] found the same tRNA genes, in the same order, but also considerable heterogeneity in the sequence and length of the intergenic parts. The authors concluded that ITS sequences are often difficult to align and therefore have little phylogenetic utility. In this study, the mitochondrial ITS sequences of twenty-five additional strains belonging to eight genotypes were retrieved from the GenBank database and analysed, allowing the identification of lineage-specific profiles.
2. Materials and Methods
Mitochondrial spacers flanked by ~120 nucleotides of the 3’ and 5’ ends of the LSU and SSU rRNA genes, respectively, were extracted from Acanthamoeba genomes available in GenBank, using the mitochondrial DNA (mtDNA) sequence of A. terricola strain Neff (U12386.1:4994–5883) [21] as a query in BLAST searches available at: [https://blast.ncbi.nlm.nih.gov] (accessed on 17 August 2025). The same search was performed on a selected set of Sequence Read Archive (SRA) data. For three strains (BCP, Linc-AP1, TN), the sequences were extracted from the available mitochondrial genomes. The remaining mitochondrial spacers analysed were those obtained by Ledee and Byers [22] (Table S1).
For the new spacers obtained, the various tRNA genes were identified by comparing with the already available sequences or by searching for the specific anticodons and modelling the secondary structures. For spacers from MG2 and MG3 species, a multiple alignment was prepared using MAFFT (L-INS-I option) and visually verified with BioEdit to ensure the correct positioning of the tRNA genes. To strengthen the analysis, 20 nt from the ends of both rRNA genes were included. In two cases, the tRNA genes for alanine (trnA) and for proline (trnP) overlapped by one or three nucleotides, and they were rearranged in the alignment as single elements. Molecular phylogenetic analyses were performed as described previously [23,24] using maximum likelihood (ML; GTR, Γ + I:4 model), distance (Neighbour-Joining, NJ, Kimura 2-P), and maximum parsimony (MP), with 1000 bootstraps.
3. Results
3.1. Acanthamoeba Species MG2/MG3 and MG1 Differ in the Arrangement of tRNA Gene Clusters
Sequences of the mitochondrial spacer flanked by the LSU and SSU ends were recovered from the genomes of twenty-three MG2 and MG3 strains belonging to six genotypes (T3, T4, T5, T11, T13, T22), as well as two MG1 strains, Acanthamoeba astronyxis (T7) and Acanthamoeba byersi (T18). For MG2 and MG3 strains, the spacer lengths range from approximately 480 nt for Acanthamoeba lenticulata (T5) to 1360 nt for Acanthamoeba palestinensis (T2), which has the particularity of having a duplicated tRNA gene for methionine (trnM) [22]. For both MG1 species, A. astronyxis and A. byersi, shorter spacers of similar length, 438 and 519 nt, respectively, were recovered.
All spacers in MG2 and MG3 strains contain the same tRNA genes (tRNA cluster 2), in the same order as previously reported [21,22]: trnI2 (isoleucine), trnA (alanine), trnP (proline), trnD (aspartic acid), and trnM (methionine). This arrangement was also observed in Acanthamoeba culbertsoni (T10), whose sequence was however excluded from the analysis due to not having sequenced gaps in the contig. In contrast, a different situation was observed for MG1 strains. A. byersi has six tRNA genes, consisting of trnM, trnY (tyrosine), trnK (lysine), trnA, trnP, and trnI2, and A. astronyxis has the same arrangement, except that it lacks trnY. Compared to MG2 and MG3, two genes (trnM and trnI2) have reversed their position, and another (trnK) appears to have replaced trnD, but at a different position. In the Neff strain, trnK is part of a group of five other tRNA genes located just upstream of the LSU rRNA gene (tRNA cluster 1) [25]. The same arrangement is found in the available mitogenomes of other T4 strains, as well as by analysing genomic data from the other genotypes studied. However, for this region as well, A. byersi and A. astronyxis exhibit a different arrangement, with the reversed position of two genes, trnQ (glutamine) and trnL1 (leucine), and the presence of trnD, which, in MG2/MG3 species, is located in the spacer (Figure 1).
Although the analysis is not exhaustive due to the limited number of strains and genotypes, it can be assumed that the arrangement of tRNA clusters 1 and 2, as observed in the Neff strain, is highly conserved in the MG2 and MG3 species. On the other hand, their rearrangement in A. byersi and A. astronyxis would allow a clear distinction between MG1 species and other Acanthamoeba, as already observed with other genetic markers. No data are available for other MG1 species. However, the order inversion between trnA and trnI2 has already been observed by Ledee and Byers [22] through preliminary results on Acanthamoeba comandoni (T9). The different arrangement of the tRNA genes could explain the difficulties encountered in completing the sequence.
3.2. Mitochondrial Spacers of Acanthamoeba Species MG2/MG3 Correlate with Lineage-Specific Profiles
In their previous study [22], Ledee and Byers compared spacers from A. terricola (Neff strain) and sixteen other strains, mainly belonging to the T4 genotype, noting high variability in length and sequence for the regions between the tRNA genes. In particular, they found that only sequences from strains known to be strictly related based on mitochondrial SSU (mtSSU) rDNA data could be reliably aligned, but only partially. The longer intergenic region, separating trnD and trnM, was often too divergent, and the spacer appeared of little phylogenetic interest. Similar high variability was also found for the additional twenty-three spacers reported here; however, differences in spacer size and region length between tRNA genes make sense when strains are ordered according to mtSSU type/subtype, particularly for T4 strains. In fact, strains of the same type/subtype have spacers and intergenic parts of similar length (Table 1).
Also, sequences of a same subtype differ from each other by a few nucleotides and/or indels, while those of distinct subtypes show greater variations but are still alignable. Furthermore, alignment is also possible for large portions of the trnD–trnM region. It is likely that the discrepancies with Ledee and Byers’s study can be explained by the larger number of sequences analysed here and the use of a different, and probably more efficient, alignment program.
Phylogenetic analysis of the spacer sequences resulted in a tree in which the T4 strains are grouped together and have the T3 and T11 strains as their sister group, consistent with results obtained with other genetic markers. It is noteworthy that the branching pattern of T4 sequences fully recognises the mtSSU subtypes as clearly distinct and strongly supported lineages, although the relationships between them are less well established (Figure 2).
In particular, several previously proposed groups [6], such as the Diamond group (T4b), Haas/BCP (Haas group) (T4a3), or Acanthamoeba lugdunensis/Linc-AP1 (T4d), are well recovered, as is the sister relationship between A. castellanii and Acanthamoeba quina.
An unexpected result is the clustering of the A. palestinensis (T2) sequence with that of genotype T22 instead of that of strain OX1 CCAP 1501/3C, because both belong to the A. palestinensis group (T2/T6 clade). The A. palestinensis spacer has a trnM duplication [22], but this is unlikely to be the cause, since the same branching was found by removing the extra trnM (not shown). A. palestinensis and OX1 differ at two or three sites in the five tRNA genes. In pairwise comparisons, the A. palestinensis spacer without the additional trnM is 68.8% similar to that of OX1, and both spacers are weakly related (~46%) to that of Acanthamoeba T22. The value between A. palestinensis and OX1 is similar to that observed between spacers of distantly related T4 subtypes (~65%), whereas between more closely related subtypes, the values are > 80%. This seems consistent with the assignment of OX1 to its own type within the A. palestinensis group, as previously suggested [13,23], and analysing more strains should resolve the relationship found here, which is likely an artifact.
3.3. RNA Editing
Most Acanthamoeba mitochondrial tRNAs undergo post-transcriptional editing to correct mismatches occurring at the acceptor stem. Modifications are limited to the first three nucleotides of the 5’ half, with the 3’ half serving as a guide and template to restore standard base pairs [26,27]. Each type of transition/transversion is possible, with pyrimidine-to-purine transversion being the most common. Ledee and Byers [22] also reported purine-to-pyrimidine editing, not observed in A. terricola, in three genes (trnP, trnD, trnM) in various strains (Acanthamoeba royreba, Diamond group, T1, T3, T2, OX1). In the present analysis, purine-to-pyrimidine editing was observed in the same genes in various other strains, as well as in trnI2 (A. lugdunensis) and trnA (Linc-Ap1, Acanthamoeba T13, A. astronyxis).
4. Discussion
Currently, Acanthamoeba strains are identified by sequencing 18S rDNA. For routine analyses, the complete sequence is too long, and the short 450-bp fragment ASA.S1 is often chosen instead. This fragment is amplified using genus-specific primers [28] and contains the 29-1 stem, which allows almost all genotypes to be distinguished. The use of mtSSU sequences is much less common, although a few previous studies have demonstrated their potential [6,8,9,10,11,29]. In various other amoebae, a fragment of the mitochondrial gene for cytochrome oxidase c subunit 1 (cox1 or COI) is increasingly being used, as it increases the ability to distinguish between closely related lineages and may be useful in unravelling cryptic species [30,31,32]. The Cox1 barcoding strategy, initially developed for animals [33,34,35], has also been applied to Acanthamoeba [20,29], appearing to be more suitable than 18S rDNA for species delimitation. Indeed, both nuclear SSU and LSU rDNA sequences seem better suited to resolving deep relationships within Acanthamoeba, i.e., to recognising genotypes, while mtSSU allows, at least in the case of the T4 genotype, less deep nodes to be disentangled as subtypes. However, groups of strains that could represent natural assemblages, i.e., species, seem to be better identified by rapidly evolving sequences such as cox1. Additional markers with similar taxonomic depths would therefore be useful, given the persistent confusion, despite previous efforts, in strain/species assignment within Acanthamoeba.
The analysis of mitochondrial rDNA spacers in Acanthamoeba performed in this study yielded lineage-specific profiles consistent with those obtained from nuclear and mitochondrial SSU rDNA sequences [6,9]. This contradicts the previously observed lack of sequence alignment and the limited utility of this region [22], likely because a larger number of strains and genotypes were studied using more powerful software. A major difference was observed between MG1 and MG2/MG3 species in the arrangement of tRNA genes (Figure 1). This should be confirmed by additional data from other MG1 strains and species. If so, such a rearrangement could provide further evidence that MG1 forms a genus-level lineage distinct from other Acanthamoeba [5,6]. No data are available regarding the mitochondrial spacer of other Acanthamoebidae, while that of the more distantly related Balamuthia mandrillaris (Centramoebida, Balamuthiidae) differs in the presence of the cytochrome oxidase subunit 2 (cox2) gene, which separates the LSU from the SSU. A cluster of five other tRNA genes is located near the SSU [36]. In other amoebozoans for which mtDNA is available, the LSU and SSU rRNA genes are either close to each other or separated by a variable number of protein-coding genes and/or tRNAs, depending on the genus/species [37]. Consequently, at present, only Acanthamoeba is characterised by a mitochondrial rDNA spacer.
The MG2 and MG3 species analysed all have the same type of mitochondrial rDNA spacer as described previously [21,22]. Its seemingly insoluble extreme heterogeneity in length and sequence can in fact be coherently correlated with different mtSSU genotypes or subtypes, both by region size profiles (Table 1) and by molecular phylogenetic analysis (Figure 2). This is particularly convincing in the case of the numerous T4 sequences, where the distribution according to the mtSSU subtype was perfectly found. The mitochondrial rDNA spacer could therefore serve as an additional marker for molecular typing and phylogenetic analysis. As an example, another T4A strain currently under study, labelled H, indistinguishable from the others by its nuclear SSU rDNA, has an mtSSU sufficiently divergent to constitute a possible new subtype. Interestingly, the strain emerges as a distinct lineage when the mitochondrial rDNA spacer is analysed (Figure 3).
The region size profile of strain H is also unique, although very similar to those of the closely related Jones and the Diamond group, varying mainly in the trnD–trnM region. For this strain, the mtSSU and spacer consistently identify a new mitochondrial subtype, although further evaluation using other markers is required to confirm this with certainty. In any case, phylogenetic analysis of spacers seems promising for identifying the different groups within T4, which is the predominant genotype in the environment and the most common in clinical samples. The T4 genotype appears to be so rich and diverse that other SSU subtypes will likely be identified in the future. Solid natural groupings can be delineated by perfect nuclear and mitochondrial matches [6,8,20], using more appropriate markers such as cox1 to unravel internal nodes, i.e., identify species. This is the case for almost all nuclear subtypes, with the exception of subtypes T4A and T4B, for which mitochondrial analysis reveals several subtypes and often mixed matches [6,29]. The mitochondrial rDNA spacer could be an additional marker, complementary to mtSSU and cox1, for analysing these relationships.
Targeting mitochondrial DNA to detect Acanthamoeba may have multiple advantages. It is estimated that growing trophozoites contain approximately 3300 mtDNA per cell [38], which is more than five times the amount of nuclear rDNA (~600 copies per cell) [39], the most commonly used target for identification. Even within cysts, the amount of mtDNA, tenfold reduced, remains important. Mitochondrial rRNA genes are easier to amplify and sequence than their nuclear counterparts, with similar results, and analysis of other genes, such as cox1, also shows good correlation with the current genotyping system [6,8,9,20,29]. The combined use of different nuclear and mitochondrial markers will certainly be useful for more reliable delineation of lineages and more accurate identification of strains.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Marciano-Cabral F. Cabral G. Acanthamoeba spp. as agents of disease in humans Clin. Microbiol. Rev.20031627330710.1128/CMR.16.2.273-307.200312692099 PMC 153146 · doi ↗ · pubmed ↗
- 2Visvesvara G.S. Moura H. Schuster F.L. Pathogenic and opportunistic free-living amoebae: Acanthamoeba spp., Balamuthia mandrillaris, Naegleria fowleri, and Sappinia diploidea FEMS Immunol. Med. Microbiol.20075012610.1111/j.1574-695X.2007.00232.x 17428307 · doi ↗ · pubmed ↗
- 3Rowbotham T.J. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae J. Clin. Pathol.1980331179118310.1136/jcp.33.12.11797451664 PMC 1146371 · doi ↗ · pubmed ↗
- 4Pussard M. Pons R. Morphologie de la paroi kystique et taxonomie du genre Acanthamoeba (Protozoa, Amoebida)Protistologica 197713557598
- 5Stothard D.R. Schroeder-Diedrich J.M. Awwad M.H. Gast R.J. Ledee D.R. Rodriguez-Zaragoza S. Dean C.L. Fuerst P.A. Byers T.J. The evolutionary history of the genus Acanthamoeba and the identification of eight new 18S r RNA gene sequence types J. Eukaryot. Microbiol.199845455410.1111/j.1550-7408.1998.tb 05068.x 9495032 PMC 7194170 · doi ↗ · pubmed ↗
- 6Corsaro D. Update on Acanthamoeba phylogeny Parasitol. Res.202011933273338 Erratum in Parasitol. Res. 2021, 120, 1927–192810.1007/s 00436-021-07102-132789533 · doi ↗ · pubmed ↗
- 7Corsaro D. Molecular evidence for greater diversity within Acanthamoeba Acta Parasitol.20257012810.1007/s 11686-025-01068-140478411 · doi ↗ · pubmed ↗
- 8Corsaro D. Venditti D. Molecular evidence for a new lineage within the Acanthamoeba T 4 genotype Parasitol. Res.20231221445145010.1007/s 00436-023-07844-037046026 · doi ↗ · pubmed ↗