Understanding Long-Term Survival in ALS: A Cohort Study on Subject Characteristics and Prognostic Factors
Elisabetta Pupillo, Elisa Bianchi, Maurizio Angelo Leone, Massimo Corbo, Massimiliano Filosto, Alessandro Padovani, Barbara Risi, Marcella Vedovello, Valentina dell’Era, Federica Cerri, Claudia Morelli, Luca Diamanti, Mauro Ceroni, Yuri Falzone, Andrea Rigamonti, Eugenio Vitelli

TL;DR
This study explores why some people with ALS live much longer than others, finding factors like younger age at diagnosis and spinal onset may contribute to longer survival.
Contribution
The study identifies clinical characteristics associated with long-term survival in ALS patients using a population-based cohort.
Findings
Long-term survivors had a median survival of 13.4 years compared to 1.9 years for non-survivors.
Long-survivors were younger at onset, had longer diagnostic delay, and were more likely to have spinal onset.
A higher proportion of males were found among long-term survivors (75%) compared to non-survivors (59%).
Abstract
Background: Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disease with variable clinical progression. While median survival is 2–4 years, 5–15% of individuals survive for longer. Methods: We conducted a retrospective, observational study using a population-based ALS register in Lombardy, Italy, to identify the clinical characteristics of long-term ALS survivors (≥10 years). Incident cases included in two periods (1998–2002 and 2008–2012) were considered. Results: A total of 828 ALS cases were included. Median survival for the entire cohort was 2.2 years (IQR 1.1–4.4). However, long-term survival was observed in 7% of individuals at 10 years, and 3% at 15 years. Long-survivors had a median survival of 13.4 years, significantly longer than the 1.9 years of non-long-survivors (IQR 1.0–3.6). Long-survivors were younger at disease onset and diagnosis, had longer diagnostic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
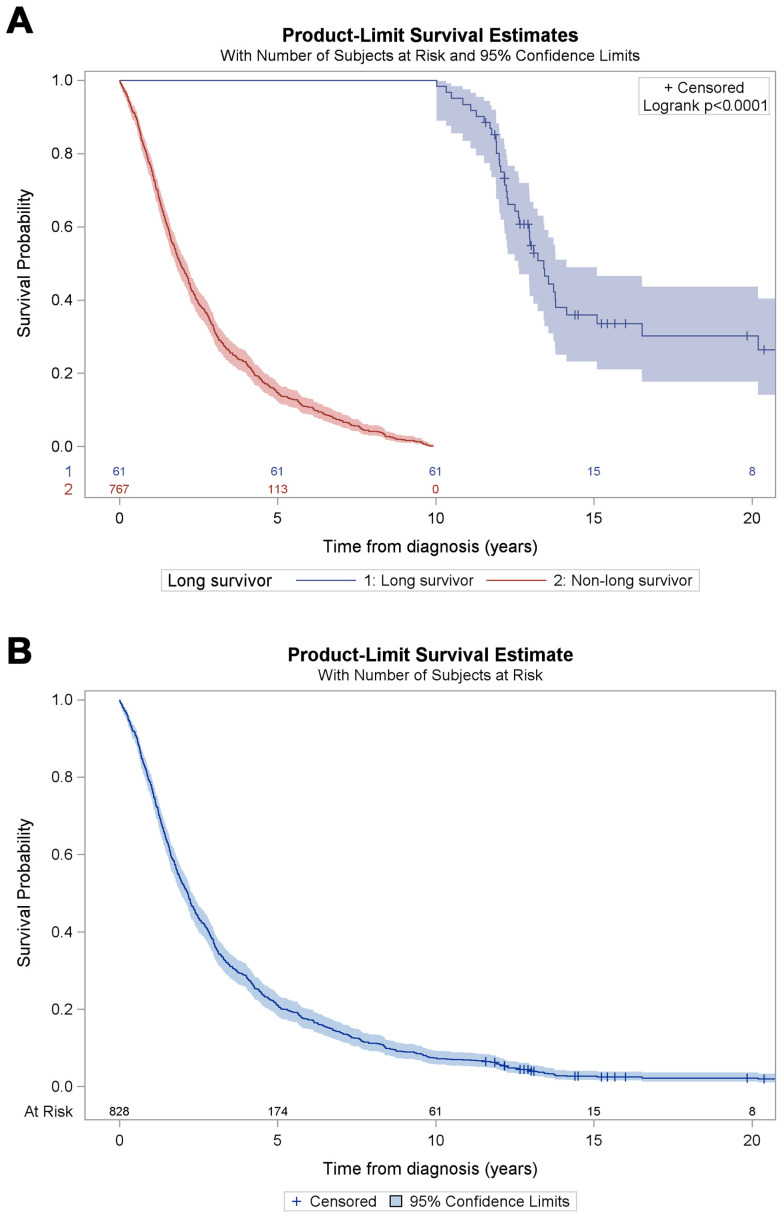 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAmyotrophic Lateral Sclerosis Research · Prion Diseases and Protein Misfolding · Biochemical Acid Research Studies
1. Introduction
A reliable prognostication of clinical course and survival in amyotrophic lateral sclerosis (ALS) seems uncertain, yet the disease is invariably deemed fatal, with median survival of 2–4 years [1,2,3]. The variability in clinical progression is traditionally thought to be related to the phenotypic heterogeneity of ALS, this being regarded as a possible predictor of longer survival, for example, in ALS with predominant upper motor neuron involvement [4,5]. Nowadays the increasing resources provided by genetic analysis are disentangling to some extent the issue, because a growing number of genes are characteristically related to slow- or fast-progressing disease, both in familial and sporadic ALS [6,7]. On the other hand, epidemiologic data provided by population-based registries are consistent with an estimate of 5–15% long-surviving ALS subjects [8], with some clinical features being recognized as good predictors of long survival: for example, spinal onset, young age of onset, and a long diagnostic delay [9,10]. Of note, there is no full agreement on the definition of long survival: in some instances, this is more than 5 years [11]; in others, it is more than 10 years.
Our aim is to investigate the clinical features of individuals with ALS who survived for 10 years or more and who were enrolled in a large population-based register of ALS patients established in Lombardy, Italy. Two five-year periods were taken into consideration, 1998–2002 and 2008–2012, in order to explore and describe clinical courses and main predictors distinguishing long-survivors from the general ALS population, and to identify possible differences over time, if any.
2. Materials and Methods
2.1. Data Collection
SLALOM (SLA LOMbardia) is a population-based register of ALS patients resident in the Lombardy region of Italy, an area of about 10 million inhabitants. SLALOM was established at the Mario Negri Institute for Pharmacological Research IRCCS, Milan in 1997. Briefly, a widespread network of local neurologists and neurophysiologists enroll every subject identified as a definite, probable, probable laboratory-supported, or possible case of ALS according to the revised El Escorial criteria. Diagnoses are refined (confirmed, changed, or rejected) during follow-up through on-site visits and detailed reviews of clinical records. Suspected ALS cases are accepted only if clinical progression is verified; such cases are subsequently allocated into a higher degree of diagnostic certainty.
Survival was calculated from date of diagnosis until date of death or of last follow-up (30 June 2024). If known, date of death was recorded by local investigators. If date of death was missing, an inquiry was sent to the municipal registry office of residency. We defined long-survivors as those surviving 10 years or more. The whole process was managed according to the General Data Protection Regulation (EU) 2016/679 (GDPR), after approval by local ethical committees.
2.2. Statistical Methods
Descriptive statistics were obtained for the entire sample, and long-survivors were compared with non-long-survivors. Continuous variables were reported as median with interquartile range (IQR) or range, categorical variables as count and percentage. Comparisons of long-survivors with non-long-survivors were carried out with the Mann–Whitney–Wilcoxon test for continuous variables; for categorical variables, the chi-square test or Fisher’s exact test was used. Survival was described for the entire sample and separately for long-survivors and non-long-survivors with Kaplan–Meier survival curves. The significance level was set at 0.05, and tests were two-tailed. Analyses were performed with the SAS statistical package (version 9.4, SAS Institute, Cary, NC, USA).
3. Results
The total number of incident ALS cases included in the analyses was 828, with 401 recruited for the period 1998–2002 and 427 for 2008–2012. Data on demographics, clinical features at diagnosis, and survival periods were all fully available, whereas data on main clinical milestones (tube feeding, mechanical ventilation) were not available in some cases (Table 1). The proportion of females/males was 45%/55%. The median age of onset was 65 years (IQR 57–72), the median age at diagnosis was 66 years (IQR 58–73), and the median diagnostic delay was 9 months (IQR 5–13). Data on other clinical characteristics are presented in Table 1. Definite, probable, and possible ALS accounted for 84.7% of cases. All the 127 subjects categorized as suspected ALS category at diagnosis were followed; in these cases, diagnosis was confirmed during follow-up.
The median estimated survival for the entire sample was 2.2 years (IQR 1.1–4.4). The cumulative survival probability was 21% at 5 years, 7% at 10 years, 3% at 15 years, and 2% at 20 years (Figure 1B).
The number of ALS subjects who survived 10 years or longer after diagnosis (long-survivors) was 61 (7%). No significant differences were identified between the two periods (1998–2002 vs. 2008–2012) (Table 1). The clinical features of the groups of individuals who survived <10 years and ≥10 years are shown in Table 1. Long-survivors were younger at both disease-onset and diagnosis, and their diagnostic delay was three months longer. Among long-survivors, 13 subjects were diagnosed as having suspected ALS according to El Escorial criteria; however, all of these individuals were subsequently categorized as having definite, possible, or probable ALS during follow-up. Long-survivors more frequently had had a spinal onset (75% vs. 59% in non-long-survivors), and a higher proportion of them were males (74% vs. 54% of non-long-survivors). Riluzole was started within one month of diagnosis in 75% of cases where data were available. The proportions of subjects with tube-feeding or non-invasive ventilation and of those treated with riluzole were not significantly different in long-survivors vs. non-long-survivors. Median survival in long-survivors was 13.4 years (12.0-not estimable), whereas it was 1.9 years (IQR 1.0–3.6) in non-long-survivors. Cumulative survival probability in long-survivors vs. non-long-survivors is shown in Figure 1A.
Among 61 long-survivors, information about tracheostomy was not available for 17 subjects. Among the other 44, tracheostomy was performed on 11 individuals, all of whom were males. The site of disease onset was spinal in nine subjects and bulbar in two. The median time of performing the procedure was 7.5 years after diagnosis. The main characteristics of the 11 long-survivors on whom tracheostomy was performed are described in Table 2. Information about genetic testing was available for 26 subjects (Table 3). Among these, 12 underwent genetic testing, and a TDP-43 mutation was identified in one subject.
4. Discussion
Prognosis of ALS and the rate of progression of the disease both encompass a noticeable variability. Population-based registers indicate that only a small proportion of ALS individuals, an estimated 5–15% of the general ALS population, are long-surviving [8,12]. Herein we describe the main clinical features of subjects who were enrolled in a population register of ALS and who survived 10 years or longer. Our study aim was to highlight predictors of long-term survival over time. Median survival in our sample was 2.2 years: 13.4 years in long-survivors and 1.9 years in non-long-survivors. The proportion of ALS long-survivors in the entire sample was 7%. Previous studies report approximately 5% to 10% of ALS patients surviving 8 years or more [6,8]. No significant difference was observed between the two examined periods, with proportions of long-survivors being, respectively, 7% and 8%. It is likely that the improvement in ALS prognosis observed in recent years [13] has not affected survival in long-survivors, but has had an effect on non-long-survivors. Improvement in the multidisciplinary supportive care provided by tertiary centers in recent years may explain this. Due to the lack of collected data on supportive care, any inference regarding its beneficial impact on patient survival remains highly speculative. In partial agreement with our own previous observations [3] and the findings of other studies [12,14,15], we found four predictors of long-term survival: lower age at onset, longer diagnostic delay, male gender, and spinal onset (see Supplementary Table S1). The median age of onset was significantly lower in long-survivors, in line with previous studies which assumed old age as a risk factor for shorter survival [9,12,14]. In our study, the diagnostic delay in long-survivors was 12 months, significantly longer than that of non-long-survivors (9 months). Males and subjects with spinal onset were more prevalent among long-survivors, probably due to overrepresentation of bulbar onset, which is associated with shorter survival [9,10,12,14] in females.
Fewer than half of subjects alive after 10 years underwent measures possibly affecting survival such as percutaneous gastrostomy, non-invasive ventilation, or tracheostomy. With regard to the remaining cases, the possibility of a slow progression of the disease must be taken into account.
A correlation between the use of riluzole and survival was not evaluated because these data were only available for 51% of sample subjects. In addition, 75% of subjects started treatment with riluzole within 1 month of the date of ALS diagnosis, and only a minority of cases reported having stopped the treatment during follow-up. In consequence, it was not feasible to evaluate whether riluzole prolonged survival in the last clinical stage of ALS [16]. The possibility of misdiagnosis cannot be completely excluded; however, all our subjects were followed by expert neurologists, and we included in this survey only suspected ALS cases who had a subsequent re-evaluation congruous with definite, possible, or probable ALS.
This study has some strengths. First of all, as a population-based study, it could provide a real-world picture of clinical course and survival of the disease. Moreover, follow-up was prolonged, with repeated visits by neurologists and thorough investigations to re-assess diagnoses that were confirmed across the entire sample. Collecting all incident ALS cases in two periods, a decade apart, could have allowed for the detection of differences in survival over time.
Nevertheless, our survey has some limitations. Unfortunately, the dates of the main clinical milestones (tube feeding, mechanical ventilation) for examined ALS individuals were not available in some cases, so these could not be related to clinical progression and survival. Most importantly, the presence/absence of tracheostomy was unknown in a proportion of long-survivors; for this reason, it was not possible to evaluate tracheostomy-free survival. Genetic testing was performed in only a very small proportion of subjects, probably only those who were admitted to ALS referral centers; consequently, no association with survival could be evaluated.
In conclusion, it is likely that long-term ALS survival results from a complex interplay of clinical factors, genetic variations, and the intrinsic rate of motor neuron degeneration. The observation of cases exhibiting prolonged survival independent of respiratory and nutritional support suggests the existence of unknown genetic and environmental modifiers. Targeted research into such long-survivors offers a promising avenue for unravelling the biological basis of slow ALS progression. In addition, a more reliable prognostication of ALS could favorably influence ALS care and economic resource allocation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Goutman S.A. Hardiman O. Al-Chalabi A. ChióA. Savelieff M.G. Kiernan M.C. Feldman E.L. Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis Lancet Neurol.20222148049310.1016/S 1474-4422(21)00465-835334233 PMC 9513753 · doi ↗ · pubmed ↗
- 2Raggi A. Monasta L. Beghi E. Caso V. Castelpietra G. Mondello S. Giussani G. Logroscino G. Magnani F.G. Piccininni M. Incidence, prevalence and disability associated with neurological disorders in Italy between 1990 and 2019: An analysis based on the Global Burden of Disease Study 2019 J. Neurol.20222692080209810.1007/s 00415-021-10774-534498172 PMC 9938710 · doi ↗ · pubmed ↗
- 3Bianchi E. Pupillo E. De Feudis A. Enia G. Vitelli E. Beghi E. Trends in survival of ALS from a population-based registry Amyotroph. Lateral Scler. Front. Degener.20222334435210.1080/21678421.2021.200416734818115 · doi ↗ · pubmed ↗
- 4Agosta F. Spinelli E.G. Riva N. Fontana A. Basaia S. Canu E. Castelnovo V. Falzone Y. Carrera P. Comi G. Survival prediction models in motor neuron disease Eur. J. Neurol.2019261143115210.1111/ene.1395730920076 · doi ↗ · pubmed ↗
- 5Sabatelli M. Zollino M. Luigetti M. Del Grande A. Lattante S. Marangi G. Monaco M.L. Madia F. Meleo E. Bisogni G. Uncovering amyotrophic lateral sclerosis phenotypes: Clinical features and long-term follow-up of upper motor neuron-dominant ALS Amyotroph. Lateral Scler.20111227828210.3109/17482968.2011.58084921702734 · doi ↗ · pubmed ↗
- 6Leighton D.J. Ansari M. Newton J. Parry D. Cleary E. Colville S. Stephenson L. Larraz J. Johnson M. Beswick E. Genotype-phenotype characterisation of long survivors with motor neuron disease in Scotland J. Neurol.20232701702171210.1007/s 00415-022-11505-036515702 PMC 9971124 · doi ↗ · pubmed ↗
- 7Waller R. Bury J.J. Appleby-Mallinder C. Wyles M. Loxley G. Babel A. Shekari S. Kazoka M. Wollff H. Al-Chalabi A. Establishing m RNA and micro RNA interactions driving disease heterogeneity in amyotrophic lateral sclerosis patient survival Brain Commun.20236 fcad 33110.1093/braincomms/fcad 33138162899 PMC 10754318 · doi ↗ · pubmed ↗
- 8Hardiman O. Al-Chalabi A. Brayne C. Beghi E. Berg L.H.v.D. Chio A. Martin S. Logroscino G. Rooney J. The changing picture of amyotrophic lateral sclerosis: Lessons from European registers J. Neurol. Neurosurg. Psychiatry 20178855756310.1136/jnnp-2016-31449528285264 · doi ↗ · pubmed ↗