Uncovering the PML::RARA Fusion in Cytogenetically Cryptic and FISH-Negative Acute Promyelocytic Leukemia—A Case Report and Comprehensive Literature Review
Busra N. Delikkaya, Jaime Eberle-Singh, Arianna B. Morton, Jerald Z. Gong, Jinglan Liu

TL;DR
This paper reports a rare case of acute promyelocytic leukemia where the genetic marker was missed by standard tests but detected later with molecular methods.
Contribution
The paper highlights the importance of molecular testing in diagnosing rare, cytogenetically cryptic APL cases missed by standard FISH and cytogenetics.
Findings
qRT-PCR confirmed the presence of PML::RARA fusion in a FISH-negative APL case.
A review of 34 similar cases showed RT-PCR reliably detects cryptic fusions.
Standard diagnostic methods often miss rare APL cases, emphasizing the need for molecular testing.
Abstract
The PML::RARA fusion resulting from t(15;17) is the genetic hallmark of acute promyelocytic leukemia (APL), typically detected by cytogenetics and/or fluorescence in situ hybridization (FISH) studies. Rarely, APL patients present with normal cytogenetics and FISH findings, complicating diagnosis and delaying life-saving therapy. We report a 23-year-old male with clinical, morphologic and immunophenotypic features consistent with APL but negative for FISH studies. Despite prompt initiation of all-trans retinoic acid (ATRA) based on clinical suspicion, the patient succumbed to intracranial hemorrhage. Quantitative reverse transcriptase PCR (qRT-PCR) confirmed a long isoform PML::RARA fusion. A review of 34 published cytogenetics- and FISH-negative cases since 1995 demonstrates that RT-PCR-based methods reliably detect cryptic fusions. While advanced genomic approaches may identify these…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
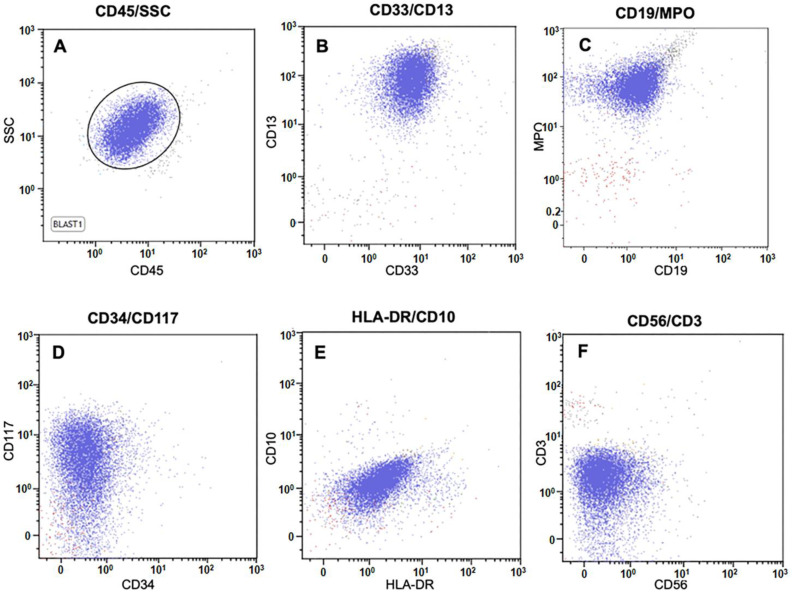 Figure 1
Figure 1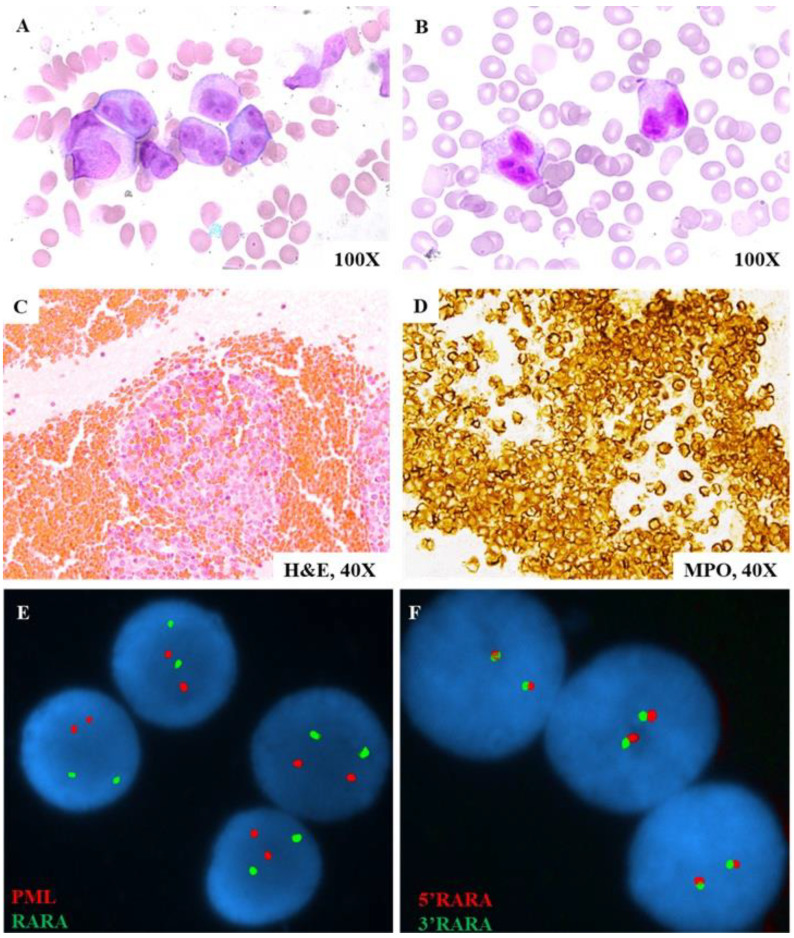 Figure 2
Figure 2| No. | Age/ | WBC (109/L) | Hb (g/dL) | Plt (109/L) | Pro-myelocytes | Immuno-phenotype | Coagulation Tests | DIC at Dx | Reason for Presentation | Karyogram | FISH # | RT-PCR * | Treatment | Survival (Months) | CR | PMID |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 39/F | 242.2 | 8.8 | 20 | Yes 1 | CD13+, CD33+, MPO+, HLA- DR+; CD34− | PT↑, aPTT↑ | Yes | History of CML with persistent fever, malaise, and weight loss | 46,XX,t(9;22)[20] | ND | S (bcr3) | IDA | No (5) | Yes | 7736444 [ |
| 2 | 25/F | 2.9 | 8.3 | 9 | Yes | CD33+; HLA-DR− | PT↑, FDP↑, | Yes | Purpura and nasal bleeding | 46,XX,del(9)(q22)[20] | ND | L (bcr1) | IDA | No (<1) | No | 10484977 [ |
| 3 | N/A | N/A | N/A | N/A | Yes | N/A | N/A | N/A | N/A | 45,XY,add(2)(q37),−7,add(9)(p22)/45,idem,add(10)(p14) | ND | L (bcr1) | N/A | N/A | N/A | 10942371 [ |
| 4 | N/A | N/A | N/A | N/A | Yes | N/A | N/A | N/A | N/A | 46,XY[20] | ND | L (bcr1) | N/A | N/A | N/A | 10942371 [ |
| 5 | N/A | N/A | N/A | N/A | Yes | N/A | N/A | N/A | N/A | 46,XX[20] | ND | Positive (unclear variant) | N/A | N/A | N/A | 10942371 [ |
| 6 | N/A | N/A | N/A | N/A | Yes | N/A | N/A | N/A | N/A | 46,XX[20] | ND | S (bcr3) | N/A | N/A | N/A | 10942371 [ |
| 7 | N/A | N/A | N/A | N/A | Yes | N/A | N/A | N/A | N/A | 46,XY[29] | ND | Positive (unclear variant) | N/A | N/A | N/A | 10942371 [ |
| 8 | N/A | N/A | N/A | N/A | Yes | N/A | N/A | N/A | N/A | 46,XY[20] | ND | V (bcr2) | N/A | N/A | N/A | 10942371 [ |
| 9 | 48/F | 14.93 | 8.8 | 36 | Yes | CD13+, CD33+, MPO+, CD56+; HLA-DR−, CD34− | PT↑, D-dimer↑, fibrinogen↓, AT (nl), aPTT (nl) | No | Vaginal bleeding, history of uterine myoma | 47,XY,+8[14]/46,XX[2] | ND | S (bcr3) | IDA | Yes (>1) | Yes | 16797070 [ |
| 10 | 14/M | N/A | N/A | N/A | Yes/hypo-cellular | Confirmed APL, WHO criteria | N/A | N/A | N/A | 46,XY[20] | ND | S (bcr3) | IDA | No (10) | Yes | 17943164 [ |
| 11 | 63/F | N/A | N/A | N/A | Yes | Confirmed APL, WHO criteria | N/A | N/A | N/A | 46,XX[20] | ND | S (bcr3) | IDA | No (6) | Yes | 17943164 [ |
| 12 | 44/F | 1.5 | 12.6 | 49 | Yes | CD13+, CD33+, MPO+; HLA-DR−, CD34− | N/A | N/A | Immature cells in peripheral blood | 46,XX,i(17)(q10)[12]/46,XX[8] | ND | L (bcr1) | IDA | Yes (>1) | Yes | 18294238 [ |
| 13 | 50/F | 101.73 | 9.9 | 16 | Yes | CD13+, CD33+, CD117+, CD7+, MPO+; HLA-DR−, CD34− | PT↑, FDP↑, fibrinogen↓, | No | Cough, fever, and general weakness for a month | 47,XY,+8[19]/46,XX[1] | ND | S (bcr3) | IDA | Yes (>5) | Yes | 19893344 [ |
| 14 | 39/F | 16.9 | 7.6 | 174 | Yes | CD13+, CD33+, MPO+; HLA-DR−, CD34− | fibrinogen↓, FDP↑, D-dimer↑, PT (nl), aPTT (nl) | No | Two weeks of fatigue, five days of high fever and irregular uterine bleeding | 46,XX,7q[7]+/46,XX[8] | ND | L (bcr1) | ATRA | Yes (>2) | Yes 2 | 19162322 [ |
| 15 | 26/F | 0.6 | 7.7 | 155 | Yes | CD13+, CD33+, CD117+; HLA-DR−, CD34− | PT↑, INR↑, | No | Fever, pallor and bleeding gums | del(5q) | ND | S (bcr 3) | IDA | Yes (>8) | Yes | 19224461 [ |
| 16 | 33/F | 1.39 | 7.1 | 9.8 | Yes | CD13+, CD33+, CD117+; HLA-DR−, CD34− | Normal | No | Fever and fatigue | Normal | ND | S (bcr 3) | ATO | Yes (>12) | Yes | 19224461 [ |
| 17 | 46/M | 63.8 | 6.6 | 146 | Yes | CD13+, CD33+, MPO+; CD117−, HLA-DR−, CD34− | PT↑, aPTT↑, INR↑ | No | Three weeks of fatigue and intermittent fever | del(19p13), del(12q24.1), del(5) | ND | L (bcr1) | IDA | Yes (>8) | Yes | 19224461 [ |
| 18 | 18/M | 16.5 | 9.5 | 37 | Yes | CD13+, CD33+, CD45+, CD117+; HLA-DR−, CD34− | N/A | No | Hematuria and hematochezia | 46,XY[20] | ND | L (bcr1), V (bcr2) | ATRA | Yes (>1) | N/A | 21156244 [ |
| 19 | 46/M | 1.7 | 10.6 | 18 | Yes | CD13+, CD33+, MPO+; HLA-DR−, CD34− | PT↑, INR↑, | No | Gengival bleeding | 92,XXYY[13]/46,XY[7] | ND | S (bcr3) | IDA | Yes (>14) | Yes | 20417966 [ |
| 20 | 24/M | 64.3 | N/A | N/A | Microgranular variant | CD13+, CD33+, CD117+; CD45+, CD34+; HLA-DR− | N/A | Yes | Suboccipital headaches, nausea, photophobia, gingival bleeding, debilitating nuchal rigidity, and low-grade fevers | 46,XY[20] | ND | Positive (unclear variant) | ATRA | Yes (>2) | Yes | 22018276 [ |
| 21 | 23/M | 3.94 | 12 | 36 | Yes | CD13+, CD33+; CD34− | N/A | N/A | Ecchymosis and thrombocytopenia | 47,XX,+8[19]/46,XY[1] | ND | L (bcr1) | ATRA + Chemo | n/a | Yes | 22402611 [ |
| 22 | 57/M | 6.82 | 8.9 | 40 | Yes | CD13+, CD33+, CD117+; HLA-DR−, CD34− | N/A | N/A | Oral ulcer, odynophagia, | 46,XY[20] | ND | L (bcr1) | ATRA | Yes (>5) | Yes | 23370423 [ |
| 23 | 67/M | 1.9 | 10.8 | 89 | Yes | MPO+; CD34−, HLA-DR− | PTT↓, fibrinogen↓, INR↑, D-dimer↑ | Yes | Easy bruising, fatigue, pancytopenia | 46,XY[20] | ND | Positive (unclear variant) | ATRA | N/A | N/A | 25580502 [ |
| 24 | 17/M | 9.9 | n/a | n/a | Yes | CD13+, CD33+, CD117+; HLA-DR−, CD34−, CD11b− | N/A | Yes | gum bleeding, multiple ecchymoses, abdominal pain, and fever | 46,XY[20] | ND | L (bcr1) | ATRA | Yes (>24) | Yes | 24561214 [ |
| 25 | 25/M | 5.2 | 11.6 | 30 | Yes | CD117+, CD64+, CD123+, CD13+, MPO+; CD34−, HLA-DR− | PT↑, INR↑, TT↑ | N/A | Gum bleeding for over 20 days | 46,XY[20] | ND | S (bcr3) | ATRA | Yes (>2) | Yes | 27995890 [ |
| 26 | 66/M | 2.95 | 78 | 7 | No 3 | CD7+, CD13+, CD33+, CD34+, Cd38+, CD117+, CD38, HLA-DR+, MPO+ | PT↑, fibrinogen↑, | N/A | Petechiae or bruises on the lower limbs that lasted for half a month with intermittent fever and coughing | 46,XY[20] | ND | V (atypical) 4 | IDA | Yes (>7) | Yes | 31959056 [ |
| 27 | 57/F | n/a | 9.7 | 57 | Microgranular variant | CD13+, CD33+, CD34+, CD117+, MPO+, CD2+; HLA-DR−, CD11b−, | fibrinogen↓ | No | Bruising and gingival bleeding over a two-week period | 46,XX[20] | ND | L (bcr1) | ATRA | Yes (>1) | Yes | 31809670 [ |
| 28 | 17/M | 48.4 | 8.3 | 14 | Yes | CD13+, CD33+, CD38+, CD45+, CD64+, CD117+, HLA-DR+ (small subset), MPO +; CD34− | fibrinogen↓, | Yes | Unresponsive during a seizure after two-day history of nausea, blood-tinged vomiting, lethargy, and right-sided weakness | 46,XY[20] | ND | Positive (unclear variant) | ATRA | N/A | Yes | 32366568 [ |
| 29 | 12/F | 22.5 | 7.4 | 16 | Yes | CD33+, CD34+, MPO+, CD117+, HLA-DR+; CD11b− | fibrinogen↓, PT↑, D-dimer (nl) | Yes | Multiple ecchymoses | 46,XX[20] | ND | S (bcr3) | ATRA | Yes (>26) | Yes | 32909480 [ |
| 30 | 56/F | 0.7 | 8 | 96 | Yes | CD13+, CD33+, CD117+, CD34+, MPO+, CD56+; HLA DR− | N/A | No | Generalized pruritis for 1 month, fever and headache | 46,XX[20] | ND | L (bcr1) | ATRA | No (<1) | No | 33851647 [ |
| 31 | 54/M | 1.6 | 9.4 | 69 | Yes | MPO+, CD117+; CD34−, HLA-DR− | N/A | N/A | Left leg swelling and a left femoral vein thrombosis, syncopal episode | 46,XY[20] | ND | L (bcr1) | ATRA | N/A | Yes | 35572917 [ |
| 32 | 27/M | 4.67 | 15 | 144 | Yes | CD13+, CD33+, MPO+; CD34−, HLA-DR− | PT↑, D-dimer↓, aPTT (nl), fibrinogen (nl), | no | Imaging findings of marrow heterogeneity in T12 during L4–L5 disk herniation work-up | 46,XY[20] | ND | L (bcr1) | ATRA | Yes (n/a) | Yes | 37685882 [ |
| 33 | 32/F | 1.57 | 8.1 | 68 | Yes | CD2+, CD13+, CD33+, CD117+, MPO+ | PT↑, fibrinogen↓, D-dimer↑, aPTT (nl), | Yes | Several weeks of fatigue and easy bruising | 46,XX[20] | ND | S (bcr3) | ATRA | Yes (n/a) | Yes | 37685882 [ |
| 34 & | 23/M | 80.1 | 4.8 | 22 | Yes | CD13+, CD33+, CD38+, CD64+, CD117+, MPO+; HLA-DR−, D34−, | PT↑, INR↑, PTT (nl) | N/A | Intracranial hemorrhage, 2 weeks post dental cleaning for pain | N/A | ND | L (bcr1) | ATRA | No (<1) | No | N/A |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRetinoids in leukemia and cellular processes · Acute Myeloid Leukemia Research · Protein Degradation and Inhibitors
1. Introduction
Acute promyelocytic leukemia (APL), with approximately 800 new cases annually, accounts for approximately 10% of adult myeloid leukemia in the United States [1]. Morphologically, it is characterized by immature hypergranular promyelocytes with abundant cytoplasm, irregular nuclei, fine azurophilic granules, and Auer rods in bone marrow and/or peripheral blood. Immunophenotyping typically shows expression of CD13, CD33, CD117, and MPO, with absent or low expression of CD34, HLA-DR, and markers of granulocytic differentiation [2,3].
The cytogenetic hallmark of APL is the reciprocal translocation t(15;17)(q22;q21) involving the PML gene on 15q24 and the RARA gene on 17q21. This fusion blocks myeloid maturation at the promyelocytic stage but generally confers a favorable prognosis due to sensitivity to all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) [2,3,4]. Approximately 90% of patients carry the balanced translocation, while 6% have complex rearrangements and 0.7% have cryptic translocations; and all result in the oncogenic PML::RARA fusion, typically detectable by conventional karyotyping or fluorescence in situ hybridization (FISH) using a PML/RARA dual-fusion probe. The remaining 1~2% of cases involve non-PML fusion partners for RARA, which can be identified by karyotyping or by FISH with a RARA break-apart probe [5].
While karyotyping and FISH remain standard-of-care diagnostic approaches, RT-PCR provides greater resolution by defining fusion isoforms and is primarily applied to monitor minimal residual disease. Due to different breakpoints in the PML and RARA genes, three major chimeric isoforms (bcr1, bcr2, and bcr3) and more than 30 atypical isoforms of the PML::RARA fusion have been identified. With the exception of a few non-PML fusions (e.g., ZBTB16/PLZF::RARA), most patients respond well to ATRA- and ATO-based therapy, underscoring the importance of timely and accurate diagnosis [3,4]. Exceptionally rare cases of APL with classic clinical and morphological features but negative results by cytogenetics and FISH have been reported. In these patients, PML::RARA fusion transcripts can still be identified by RT-PCR, and excellent outcomes are achieved when treatment is initiated promptly [6,7].
Herein, we describe a new APL case with typical morphology and immunophenotype but negative cytogenetics and FISH results, in which a long PML::RARA transcript was identified by RT-PCR. We also review the clinicopathologic features of 34 similar cases reported since 1995 and discuss alternative molecular and genomic approaches that may aid diagnosis. Of note, APL cases with cytogenetic abnormalities involving chromosome 15 or 17, or with abnormal FISH signals, even if atypical, were excluded. Cases with RARB, RARG, or some MLL and NPM1 rearrangements which can clinically and morphologically mimic typical APL with RARA rearrangements, are not addressed in this manuscript, as they have been thoroughly reviewed elsewhere [3,4]. Additionally, cases with incidental t(15;17) translocations lacking clinical symptoms or molecular evidence of gene fusions were also excluded.
2. Materials and Methods
2.1. Clinical and Pathological Examination
Clinical data, including history, presentation, laboratory findings, management, and disease course, were obtained from chart review. Peripheral blood smears were prepared and examined using the Wright-Giemsa method.
2.2. Flow Cytometry
Expression of cell surface and intracellular markers was assessed by flow cytometric immunophenotyping using the FACSLyric cytometer (Becton Dickinson, San Jose, CA, USA). A six-color panel including FITC, PE, PerCP-Cy5.5, PE-Cy7, APC, and APC-H7-conjugated antibodies was employed to evaluate lineage-specific antigens. Intracellular staining was performed using the Fix & Perm Cell Permeabilization Kit (Caltag Medsystems Ltd., Buckingham, UK), following surface staining. Data were analyzed using FACSLyric Suite and Beckman Coulter Kaluza analysis software version 1.2 with CD45 versus side scatter gating.
2.3. Conventional Cytogenetic and Interphase Fluorescence In Situ Hybridization (FISH) Studies
Standard direct harvest and 24 h unstimulated cell culture were conducted with peripheral blood cells. Interphase FISH was carried out using dual-color dual-fusion (DCDF) PML/RARA translocation and RARA break-apart (BAP) probe sets (CytoTest Inc., Rockville, MD, USA). A one-hour STAT FISH was performed according to the manufacturer’s instruction. Karyotyping and FISH images capture and analysis were conducted with the ASI system (Applied Spectrum Imaging, Carlsbad, CA, USA). Findings were analyzed and interpreted according to the International System for Human Cytogenetic Nomenclature (ISCN) 2020.
2.4. Literature Review
A PubMed search was performed using the keywords “acute promyelocytic leukemia,” “atypical,” “cryptic,” “cytogenetic negative,” and “FISH negative.” Articles published in both English and non-English were considered. No specific publication date range was applied to ensure comprehensive coverage of all available cases. The literature search was conducted independently by two researchers, and the senior author assessed all cases and finalized the list for inclusion. Reviews, case reports, and original research articles were all included. Clinical, cytogenetic, or molecular features of selected cases with atypical or cryptic acute promyelocytic leukemia were carefully reviewed. Based on the available data, only cases with typical normal DCDF PML::RARA FISH signals, typical normal RARA BAP signals, normal karyograms, or abnormal karyograms not involving chromosomes 15 and 17 were included.
3. Results
3.1. Case Study
A 23-year-old male presented to the emergency department with left upper extremity weakness, occurring two days after undergoing a dental cleaning. Imaging revealed an intracranial hemorrhage, necessitating an urgent decompressive hemicraniectomy. Flow cytometry analysis demonstrated a cell population characterized by CD4−, CD10−, CD11b−, CD11c−, CD13+, CD14−, CD16−, CD33+, CD34−, CD38+, CD56−, CD64+(dim), CD117+, CD235a−, HLA-DR−, cCD3−, cCD22−, cCD79a−, MPO+, and TdT- expression (Figure 1). Peripheral blood smear examination identified abnormal promyelocytes (Figure 2A,B). These findings were consistent with acute promyelocytic leukemia (APL). However, STAT FISH analyses for PML::RARA fusion and RARA break-apart were negative (Figure 2E,F). Despite the absence of cytogenetic and FISH evidence, a diagnosis of APL was made based on the morphology and immunophenotypic profile. Treatment with all-trans retinoic acid (ATRA) was initiated immediately. Unfortunately, the patient’s post-operative course was complicated by severe coagulopathy, pancytopenia, and multi-organ failure, ultimately resulting in death seven days after admission. Histomorphological examination of the specimen from the right-sided temporectomy revealed multiple brain parenchymal hemorrhages with dense clusters of leukemia cells that stained positive for myeloperoxidase (MPO) (Figure 2C,D). Subsequent reverse transcriptase-polymerase chain reaction (RT-PCR) analysis, performed at an external commercial laboratory, was reported a week later, confirmed the presence of a PML::RARA long fusion transcript. Thus, the diagnosis of APL was established despite the negative findings from conventional cytogenetic studies.
3.2. Literature Review
Table 1 summarizes the clinical, histomorphological, and molecular genetic profiles of 34 APL patients lacking cytogenetic or FISH evidence of t(15;17). The median age was approximately 3540 years (range 1267), with a nearly equal male-to-female distribution. Most presented with bleeding, cytopenias, and laboratory evidence of disseminated intravascular coagulation (DIC) at the time of hospital admission. Morphologically, most cases showed classic hypergranular promyelocytes. Two cases exhibited microgranular variants, one showed a mixture of classic hypergranular promyelocytes with a high proportion of microgranular promyelocytes, and one was consistent with AML-FAB M5. Although t(15;17) was not detected by cytogenetics or FISH, all patients showed PML::RARA fusion transcripts by RT-PCR. The long (bcr1) isoform was most common (14 cases), followed by the short (bcr3) isoform (12 cases). The variant (bcr2) isoform was rare, observed in 2 patients. One patient had both bcr1 and bcr2 isoforms. The remaining five patients were positive for fusion transcripts without specification of the isoforms. Additional abnormalities, including trisomy 8, del(5q), del(9q), and i(17)(q10), were observed in a minority of cases but did not affect the molecular diagnosis. Treatment strategies evolved over time. Early cohorts were managed with ATRA combined with chemotherapy, whereas more recent patients received ATRA with arsenic trioxide (ATO), with or without chemotherapy. Overall, most patients achieved hematologic remission, although early deaths still occurred, predominantly due to hemorrhagic complications (intracranial or pulmonary) and, less frequently, severe infections. Early mortality, largely due to hemorrhagic complications, remained a significant cause of treatment failure. Long-term survival data were not available, but the modern ATRA + ATO-based regimens appear to be associated with superior remission durability.
4. Discussion
APL is a rare and aggressive AML subtype defined by the PML::RARA fusion. Conventional karyotyping and FISH are first-line diagnostic tests, while RT-PCR-based assays confirm transcript isoforms and enable evaluation of minimal residual disease (MRD) during clinical follow-ups. Rarely, patients with typical clinical and histomorphological features and a favorable response to ATRA+ATO therapy lack detectable PML::RARA fusion by these assays [6,7]. We systematically reviewed 34 reported cases of cytogenetics- and FISH-negative APL published since 1995, including both English- and non-English-language reports. To the best of our knowledge, this constitutes the most comprehensive review of this rare entity to date.
Clinically, these patients closely resemble those with classic t(15;17) translocations, showing comparable hematologic profiles, immunophenotypes, and coagulopathy features (Table 1). Mechanistically, submicroscopic insertions are believed to cause the fusion in most cases [9,10,30,31,32]. These insertions often involve multiple breakpoints and are mediated by microhomology-mediated break-induced replication (MMBIR) or fork stalling and template switching (FoSTeS), resulting in the formation of the PML::RARA fusion. This represents a distinct genomic event from the conventional reciprocal t(15;17), which arises via non-allelic homologous recombination (NAHR) or breakage at palindromic AT-rich repeats (PATRRs) [33,34]. Together, these diverse mechanisms suggest that genes involved in DNA replication or repair may drive leukemogenesis even prior to fusion formation, underscoring the potentially distinct biology of cryptic APL.
Diagnostic approaches reported in the literature are summarized in Table 2, with a focus on diagnostic yield, turnaround time (TAT), potential pitfalls, and clinical applicability. Conventional karyotyping remains foundational, detecting the classic t(15;17) translocation and additional chromosomal abnormalities in over 96% of cases [5]. However, its sensitivity is reduced in atypical cases due to dependence on high-quality metaphase spreads and inability to resolve cryptic rearrangements. Nevertheless, its genome-wide scope allows detection of co-existing abnormalities with prognostic or therapeutic significance. FISH assays offer rapid, high-sensitivity detection of typical and atypical fusions (98~100% yield) when PML/RARA dual-color dual-fusion and RARA break-apart probes are used in parallel [3,32,35]. Their short TAT (3 h) makes them valuable for urgent diagnostic confirmation, particularly in life-threatening coagulopathy. Limitations include hybridization failure, signal overlap, and inability to detect very small insertions, which may account for the rare subset of cytogenetics- and FISH-negative cases. Nonetheless, their low-to-moderate cost and widespread availability ensure their continued role as frontline diagnostic tools. RT-PCR-based molecular assays provide rapid, high-resolution detection of PML::RARA isoforms and are essential for MRD monitoring, with qRT-PCR considered the gold standard [18,26]. These assays are widely available and affordable, though rare atypical breakpoints may generate false negatives [36]. Sequencing of RT-PCR products allows precise characterization of fusion variants, albeit with longer TAT and higher RNA quality requirements. Advanced genomic technologies, including array comparative genomic hybridization (aCGH/tCGH), mate-pair sequencing (Mpseq), and optical genome mapping (OGM), enable detection of cryptic structural variants across broader genomic regions [30,37]. OGM, in particular, shows promise for resolving structural rearrangements and fusion breakpoints at a medium-level resolution [9]. However, these platforms require high-quality input material and specialized expertise, and challenges remain in detecting polyploidy, mosaicism, or rearrangements in heterochromatic or highly repetitive regions. TATs are longer (minimum 35 days), costs higher, and access restricted to specialized centers. At the highest resolution, rapid NGS panels, whole-genome and whole-exome sequencing (WGS/WES) provide targeted or comprehensive detection of fusion partners and profiling of co-existing aberrations [28,37]. Their application remains largely research-focused due to high cost, bioinformatics complexity, interpretation challenges, longer TAT, and the need for variant validation.
Collectively, these methods form a complementary, hierarchical diagnostic framework (Table 3). First-line testing with karyotyping, FISH, and RT-PCR is sufficient for most cases, offering high diagnostic yield, short TAT, and wide accessibility at relatively low cost. Advanced technologies such as OGM and WGS/WES are best reserved for ambiguous cases where conventional techniques fail, as well as for expanding our understanding of APL genomics. This tiered approach balances diagnostic precision with practical feasibility, while addressing the small but clinically significant subset of cytogenetics- and FISH-negative APL.
This review is limited by the small number of reported cases and incomplete documentation of clinical and demographic data, which precludes meaningful statistical analysis. The 34 cases identified across 25 manuscripts, including the present study (Table 1), lacked consistent information on ethnicity and racial background. Thirteen studies (52%) originated from groups in China, India, Japan, and Korea, and including our patient from India, 17 patients (50%) were presumably of Asian descent. Given the limited demographic, epidemiological, and socioeconomic data, no conclusions can be drawn about the role of genetic background or environmental factors in the pathogenesis of this ultra-rare APL subset. Furthermore, all studies were retrospective in design. Most were single case reports, with a few small case series, which limits the generalizability of the findings. Potential reporting bias and heterogeneity of the available data could not be confidently assessed. Supplementary Table S1 summarizes the limitations and potential bias for each case. These cases, however, point to a possible association between cryptic APL and ethnicity/race, emphasizing the need for more detailed reporting in future cases.
Despite the small cohort size, several important questions remain. First, it is unclear whether cryptic rearrangements detected by RT-PCR-based methods are balanced or unbalanced, as unbalanced fusions may acquire novel functions that influence phenotypic outcomes [38]. Second, the mutational spectrum of co-existing primary or secondary genetic alterations is poorly defined. Experimental studies in transgenic mice suggest that cooperating mutations are essential to drive full leukemic transformation [39]. In de novo APL, co-existing mutations, including point mutations in the PML::RARA fusion gene, occur in approximately ~70% of cases and also significantly contribute to relapse or therapy resistance [40,41]. These mutations are heterogeneous but may converge functionally to drive APL initiation and confer therapy resistance [23,24]. Third, the impact of such alterations on higher-order chromosome architecture, epigenetic regulation, and transcriptional networks remains largely unknown.
Clinically, APL patients lacking cytogenetic or FISH evidence face high risk of diagnostic delay and potentially fatal early hemorrhage [28]. Empiric initiation of ATRA upon clinical suspicion remains essential, even when first-line tests are negative. Emerging approaches, including OGM, long-read sequencing, and multi-omics technologies (epigenomics, transcriptomics, metabolomics), hold promise for illustrating cryptic mechanisms and revealing therapeutic targets. Despite major therapeutic advances, early mortality of APL remains ~15%. Studying this ultra-rare subset may not only improve outcomes in cryptic APL but also yield broader insights into APL biology and leukemogenesis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mai B. Liang C. Nguyen A. Wahed A. Chen L. Cryptic Acute Promyelocytic Leukemia (APL) Presenting as Seizures in an Adolescent Ann. Clin. Lab. Sci.20205027027332366568 · pubmed ↗
- 2Li G. Wu J. Li R. Pan Y. Ma W. Xu J. Nan M. Hou L. Improvement of Early Death in Acute Promyelocytic Leukemia: A Population-Based Analysis Clin. Lymphoma Myeloma Leuk.202323 e 78e 843656721410.1016/j.clml.2022.11.005 · doi ↗ · pubmed ↗
- 3Zhang X. Sun J. Yu W. Jin J. Current views on the genetic landscape and management of variant acute promyelocytic leukemia Biomark. Res.202193310.1186/s 40364-021-00284-x 33957999 PMC 8101136 · doi ↗ · pubmed ↗
- 4Guarnera L. Ottone T. Fabiani E. Divona M. Savi A. Travaglini S. Falconi G. Panetta P. Rapanotti M.C. Voso M.T. Atypical Rearrangements in APL-Like Acute Myeloid Leukemias: Molecular Characterization and Prognosis Front. Oncol.20221287159010.3389/fonc.2022.87159035494081 PMC 9039303 · doi ↗ · pubmed ↗
- 5Gagnon M.F. Berg H.E. Meyer R.G. Sukov W.R. Van Dyke D.L. Jenkins R.B. Greipp P.T. Thorland E.C. Hoppman N.L. Xu X. Typical, atypical and cryptic t(15;17)(q 24;q 21) (PML::RARA) observed in acute promyelocytic leukemia: A retrospective review of 831 patients with concurrent chromosome and PML::RARA dual-color dual-fusion FISH studies Genes. Chromosomes Cancer 20226162963410.1002/gcc.2307035639830 · doi ↗ · pubmed ↗
- 6Kim M.J. Cho S.Y. Kim M.H. Lee J.J. Kang S.Y. Cho E.H. Marschalek R. Meyer C. FISH-negative cryptic PML-RARA rearrangement detected by long-distance polymerase chain reaction and sequencing analyses: A case study and review of the literature Cancer Genet. Cytogenet.201020327828310.1016/j.cancergencyto.2010.08.02621156244 · doi ↗ · pubmed ↗
- 7Rashidi A. Fisher S.I. FISH-negative, cytogenetically cryptic acute promyelocytic leukemia Blood Cancer J.20155 e 32010.1038/bcj.2015.4726090620 PMC 4648483 · doi ↗ · pubmed ↗
- 8Emilia G. Marasca R. Longo G. Ferrari M.G. Notohamiprodjo M. Temperani P. Sacchi S. Torelli G. Detection of PML-RAR alpha fusion transcript in Ph positive leukemia with acute promyelocytic phenotype lacking the t(15;17) cytogenetic abnormality Cancer Genet. Cytogenet.199580959910.1016/0165-4608(94)00147-47736444 · doi ↗ · pubmed ↗