ZC3H12A: A Critical Mediator of Inflammation, Tumor Immunotherapy, and Metabolic–Immune Crosstalk—Implications for Disease Treatment
Mingjun Lu, Jingwei Guo, Chenyang Wang, Bingbing Wan, Teng Ma

TL;DR
ZC3H12A is an RNA-binding protein that regulates inflammation, immune balance, and cancer treatment, offering new therapeutic potential for various diseases.
Contribution
This review highlights ZC3H12A's multifunctional roles in immune regulation, tumor therapy, and metabolic-immune crosstalk, suggesting new treatment opportunities.
Findings
ZC3H12A degrades inflammatory mRNAs like IL-6 and IL-1β and regulates immune cells.
ZC3H12A knockout improves T-cell persistence and anti-tumor efficacy in immunotherapy.
ZC3H12A influences metabolic-immune interactions and inflammation-related diseases.
Abstract
ZC3H12A is a key RNA-binding protein and ribonuclease that plays a central role in negatively regulating inflammation and maintaining immune homeostasis. It does this by degrading the mRNA of multiple inflammatory mediators (such as IL-6 and IL-1β), as well as through its deubiquitinating enzyme activity. Not only does it limit excessive immune activation by regulating innate and adaptive immune cells (e.g., macrophages and T cells), but it also exerts bidirectional effects in tumors, acting as an anti-tumor factor to inhibit angiogenesis and oncogenic signal pathways, while promoting tumor progression under specific conditions. In recent years, ZC3H12A has emerged as a critical target for tumor immunotherapy, particularly CAR-T cell therapy. Its knockout significantly enhances T-cell persistence and anti-tumor efficacy, demonstrating broad translational potential. Furthermore, ZC3H12A…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
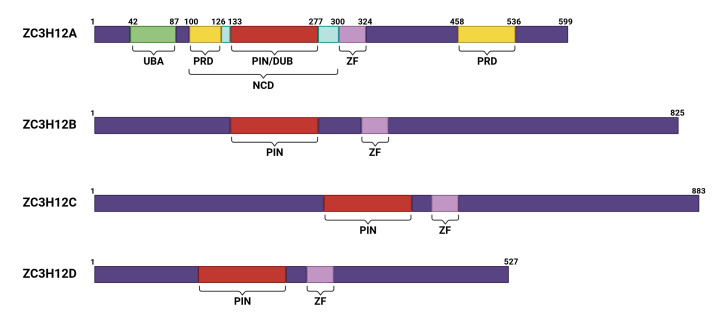 Figure 1
Figure 1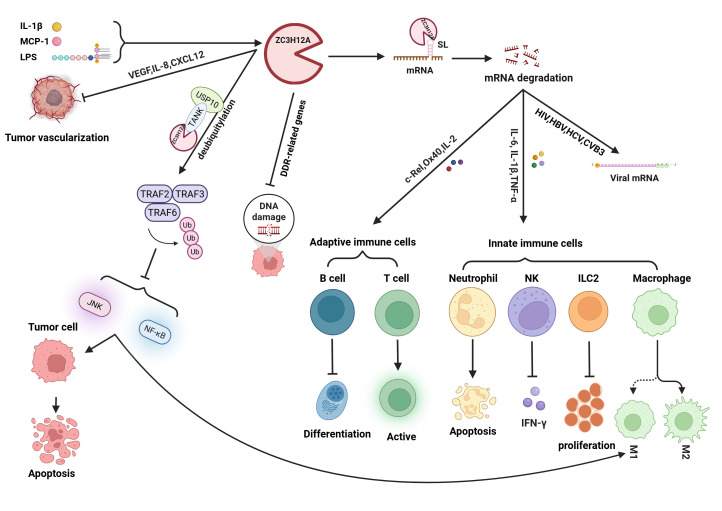 Figure 2
Figure 2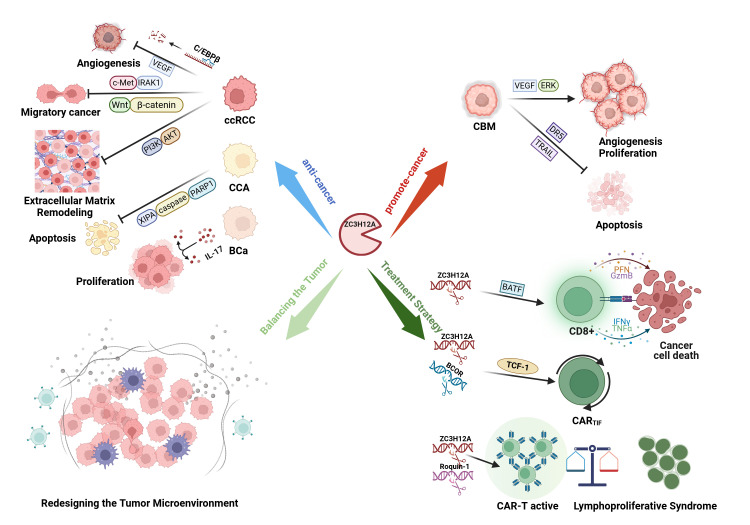 Figure 3
Figure 3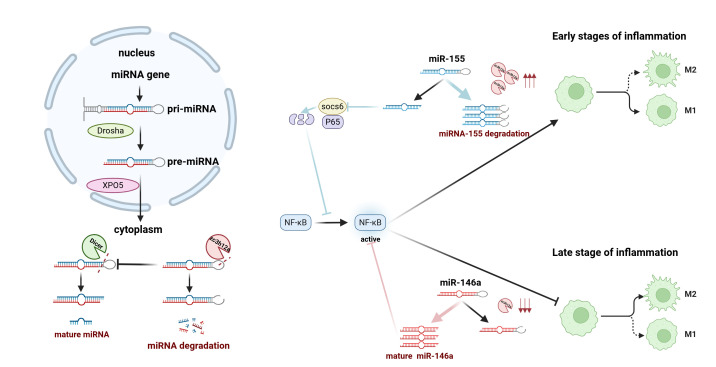 Figure 4
Figure 4- —Fundamental Research Funds for the Central Universities
- —Beijing Municipal Public Welfare Development and Reform Pilot Project for Medical Research Institutes
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdenosine and Purinergic Signaling · Immune cells in cancer · Macrophage Migration Inhibitory Factor
1. Introduction
Chronic inflammation is a critical link between intrinsic factors, such as oncogenes and tumor suppressor genes, and extrinsic factors, such as immune and stromal components, in tumor development [1]. It promotes repeated tissue damage and repair, increases mitotic errors, and facilitates the accumulation of cancer-prone cells, with epidemiological studies implicating it in 15–20% of cancers [2,3]. Inflammatory responses are governed by the dynamic regulation of gene expression in immune cells, in which RNA metabolism, especially post-transcriptional control, plays a pivotal role. Toll-like receptors (TLRs) recognize pathogens and trigger signaling that induces immune gene transcription and cytokine production [4,5]. Additionally, post-transcriptional mechanisms, mediated by RNA-binding proteins and specific nucleases, regulate mRNA stability and translation by targeting cis-elements, such as AU-rich elements and stem-loop structures in the 3′ UTR, thereby preventing excessive inflammation [6,7,8,9].
ZC3H12A, as an RNA-binding protein (RBP), plays a crucial role in regulating immune homeostasis and inflammatory factors [10]. In 2006, MCP-1-induced protein (MCPIP) was first identified in MCP-1-stimulated human monocytes as a novel protein derived from a highly induced, previously uncharacterized expressed sequence tag (EST), localized to the cell nucleus, and induced cardiomyocyte apoptosis [11,12]. Subsequent studies mapped this EST to the cDNA clone AW206332 (GenBank), identifying the gene ZC3H12A located on chromosome 1p34.3. It was proposed that ZC3H12A could bind cloned fragments of the cdh12 and cdh19 genes with DNA sequence specificity, implying a potential role as a transcriptional regulator In 2008, Liang et al. reported that lipopolysaccharide (LPS) induced the expression of ZC3H12A in macrophages [13]. They further identified a gene family comprising ZC3H12A, ZC3H12B, ZC3H12C, and ZC3H12D, all of which contain the CCCH zinc finger domain [14]. By 2009, ZC3H12A (also termed MCPIP1 or Regnase-1) was established as a ribonuclease that binds to the 3′ UTR of target mRNAs via its CCCH zinc finger domain and cleaves transcripts encoding inflammatory mediators, thereby playing an essential role in preventing autoimmunity diseases and regulating inflammation [15].
The ZC3H12A protein consists of 599 amino acids and contains multiple domains with distinct functions (Figure 1) [16]. Among these, proline-rich domains (PRDs) are located at positions 100–126 (37% proline) and 458–536 (28% proline). These non-conservative domains significantly contribute to structural stability and functional activity by regulating conformational dynamics and protein interactions [12]. ZC3H12A also contains a CCCH-type zinc finger domain (ZF). CCCH zinc finger proteins constitute only 0.8% of all zinc finger proteins and possess RNA-binding capability [17,18]. Nearly 60 such proteins have been identified in humans and mice that modulate mRNA decay and immune signaling [19]. Unlike tristetraprolin (TTP) family proteins that target AU-rich elements [20,21], the ZF in ZC3H12A recognizes stem-loop (SL) structures and facilitates RNA degradation via hydrophobic stacking [9,22]. The PIN domain constitutes the RNase active site of ZC3H12A [14,23]. Key residues (Asp141, Asp225, Asp226, and Asp244) mediate Mg^2+^-dependent RNase activity [15,23], with Asp141 being essential [24]. Intramolecular interactions enable targeted degradation of inflammatory mRNAs (e.g., IL-6 and IL-1β) (Figure 2) [25]. Additionally, ZC3H12A contains a deubiquitinating enzyme domain (DUB) that overlaps with the PIN domain. Among the five major existing DUB families, it exhibits only distant homology with enzymes from the UCH family, indicating that it belongs to a novel category of DUBs [26,27,28]. This DUB activity enables ZC3H12A to deubiquitinate TRAF2, TRAF3, and TRAF6, thereby inhibiting JNK and NF-κB signaling and reducing the production of proinflammatory cytokines [26]. Furthermore, ZC3H12A forms a complex with USP10 via the adaptor protein TANK, enhancing its ability to remove polyubiquitin chains from NEMO and TRAF6, and suppress genotoxic NF-κB activation [29]. The physiological importance of this regulatory function is underscored by the hyperactivation of NF-κB observed in ZC3H12A-deficient mice [29,30].
Early studies primarily focused on describing the multifunctionality of ZC3H12A, including PRD-mediated protein interactions, ZF-dependent mRNA binding, PIN-driven RNA degradation, and DUB-mediated NF-κB regulation. These properties highlight its central role in post-transcriptional and post-translational regulation of inflammation and immune homeostasis [31]. However, recent research has been delving into the central role of ZC3H12A within complex disease networks—such as cancer, autoimmune diseases, and viral infections—while exploring its translational potential as a therapeutic target. This review will focus on these recent advances, emphasizing ZC3H12A’s integrative functions in disease mechanisms and its application prospects in targeted therapies.
2. ZC3H12A as the “Master Switch” of Immune Homeostasis
ZC3H12A is a key protein with RNA-binding activity and ribonuclease function, performing extensive regulatory roles in the immune system that extend beyond conventional mRNA degradation [32]. It plays a central role in the cross-network between innate and adaptive immunity by precisely regulating the expression of inflammatory factors and immune-related genes at the post-transcriptional level, maintaining immune homeostasis through multiple mechanisms (Figure 2).
2.1. Regulatory Functions in Innate Immunity
Innate immune cells, such as macrophages, neutrophils, and natural killer (NK) cells, recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) through pattern recognition receptors (PRRs) expressed on their surface or intracellularly [33]. In macrophages, ZC3H12A exerts a crucial “braking” function by degrading mRNA of key inflammatory factors such as IL-6, IL-12b, and TNF-α, thereby limiting excessive activation of M1 polarization. Studies indicate that ZC3H12A deficiency significantly enhances mRNA stability of these cytokines, triggering cytokine storms and systemic inflammation [15,34,35,36]. Concurrently, ZC3H12A plays a crucial role in tissue repair and inflammation resolution by suppressing NF-κB and JNK/c-Myc signaling pathway activity, thereby promoting macrophage polarization toward the M2 type [14,15,37]. Notably, ZC3H12A expression itself is tightly regulated. Inflammatory mediators such as IL-1 and IL-17 can induce ZC3H12A expression by releasing its mRNA translation inhibition, forming a negative feedback regulatory loop [37,38,39,40]. In neutrophils, ZC3H12A participates in regulating the apoptosis process, influencing the resolution of inflammatory responses. In inflammatory bowel disease (IBD) models, its expression levels are closely correlated with pathological indicators such as myeloperoxidase (MPO) activity, reactive oxygen species (ROS) production, and cell migration [41,42]. Clinical observations indicate that elevated expression of ZC3H12A in neutrophils of patients with acute infections correlates with the severity of damage to target organs such as the liver [43]. In NK cells, deficiency of ZC3H12A enhances OCT2/IκBζ–NF-κB complex-mediated transcription of IFN-γ, thereby strengthening antitumor immune responses [44,45,46,47]. Recent studies have revealed that ZC3H12A also serves as a key molecule regulating the pro-fibrotic function of type II innate lymphoid cells (ILC2s). By degrading the mRNA of transcription factors Gata3 and Egr1, it indirectly suppresses the production of pro-fibrotic cytokines such as IL-5 and IL-13, thereby limiting the progression of idiopathic pulmonary fibrosis (IPF) [48]. Further research has revealed a novel upstream regulatory mechanism: the transcription factor Mef2d acts as a “brake” on ZC3H12A by binding to the ZC3H12A locus and suppressing its transcription. When Mef2d is absent, ZC3H12A expression increases, leading to excessive degradation of IL-33 receptor (ST2) and Gata3 mRNA, ultimately weakening the activation and fibrogenic capacity of ILC2s. This regulatory axis—from Mef2d to ZC3H12A and downstream fibrotic programs—provides a novel perspective on the immunopathological mechanisms of fibrotic diseases and suggests this pathway as a potential therapeutic intervention target [49].
2.2. “Brakes and Accelerator” of Adaptive Immunity
Within the adaptive immune system, ZC3H12A exhibits complex bidirectional regulatory properties, functioning both as a “brake” to prevent excessive immune activation and as an “accelerator” to promote effective immune responses under specific conditions. In B cells, ZC3H12A deletion enhances proliferation and class-switch recombination, resulting in severe immunopathology, indicating its fundamental role in preventing B cell-dependent autoimmunity [50]. In T cells, ZC3H12A prevents the generation of abnormal effector CD4 + T cells by targeting mRNAs encoding transcription factors (c-Rel), surface molecules (Ox40), and cytokines (IL-2) [51]. T-cell receptor (TCR) signaling dynamically regulates ZC3H12A through Malt1-mediated cleavage, facilitating full T-cell activation [51]. Furthermore, the regulatory network formed between ZC3H12A and the RNA-binding protein Roquin is of particular interest. These two proteins physically interact with each other, and disrupting this interaction enhances the effector function and tumor accumulation capacity of tumor-specific T cells [52]. Roquin deficiency primarily impacts Tfh cell differentiation and humoral immunity, whereas ZC3H12A deficiency more readily induces Th17-mediated inflammatory responses. Further knockdown of ZC3H12A in a Roquin-deficient background continues to enhance Th17 differentiation, indicating that the two proteins have non-overlapping regulatory functions [53]. This indicates that ZC3H12A may serve as a potential target for regulating T cell activity. Currently, an anti-degradation mutant of ZC3H12A has been developed. However, it suppresses TCR-β expression and signaling, thereby impairing positive selection in the thymus and increasing susceptibility of thymocytes to apoptosis. Therefore, when utilizing ZC3H12A to regulate T cell activity, caution is warranted regarding the potential risk of severe lymphopenia [54].
ZC3H12A plays a key role in regulating immune homeostasis by modulating the initiation and resolution of inflammatory responses in innate immune cells, as well as balancing the intensity and specificity of immune responses in adaptive immune cells. Understanding these regulatory mechanisms improves our understanding of the pathogenesis of immune-related diseases.
3. Bidirectional Regulation of ZC3H12A in Tumors: From “Tumor Suppressor Gene” to “Therapeutic Lever”
ZC3H12A contributes to antitumor immunity and influences multiple biological properties of tumor cells. As a central regulator within the cytokine network, it participates in tumorigenesis and progression through mechanisms including gene expression regulation and miRNA modulation and is closely associated with the homeostasis of the tumor microenvironment (Figure 3) [31,55].
3.1. The Intrinsic “Environment-Dependent” Role of Tumor Cells
ZC3H12A exhibits highly context-dependent roles in cancer biology, functioning as either a tumor suppressor or an oncogene depending on the specific cellular signaling environment. Its varying activity across different malignant tumors demonstrates this duality.
In most cancers, ZC3H12A exerts potent tumor suppressing effects by targeting key signaling pathways. In clear cell renal cell carcinoma (ccRCC), angiogenesis is a critical driver of cancer progression [56]. ZC3H12A inhibits angiogenesis in ccRCC by degrading transcription factor C/EBPβ and angiogenic factors such as VEGF and IL-8 through its RNase activity [57,58,59,60]. Simultaneously, its function is also achieved through negative regulation of the c-Met/IRAK1 signaling axis. When ZC3H12A expression is downregulated—particularly in TKI-resistant states—IRAK1 activity is enhanced, accelerating ZC3H12A protein degradation and releasing inhibition of the c-Met receptor. This process drives epithelial–mesenchymal transition, angiogenesis, and stemness maintenance [61,62,63]. Its mechanism of action is further reflected in the regulation of the c-Met/CD44 signaling axis: ZC3H12A exerts its effects by negatively regulating the stem cell marker CD44 (a key co-receptor for c-Met). Deletion of ZC3H12A elevates CD44 levels, enhances c-Met phosphorylation, and activates downstream pathways, thereby driving tumorigenesis and stemness maintenance. This highlights its function as deeply embedded within the ccRCC-specific signaling network [64]. Additionally, ZC3H12A suppresses the development of clear cell renal cell carcinoma (ccRCC) by inhibiting the Wnt/β-catenin signaling pathway. It achieves this by degrading specific miRNAs (miR-519a/b-3p and miR-520c-3p), thereby releasing their suppression of Wnt pathway inhibitors (SFRP4, KREMEN1). This stabilizes the β-catenin degradation complex, ultimately inhibiting β-catenin nuclear translocation and epithelial–mesenchymal transition (EMT) [65]. Its effects also extend to regulating the PI3K/AKT signaling axis and extracellular matrix remodeling by positively modulating PTEN and maintaining RECK and TIMP3 levels, which collectively suppress excessive PI3K/AKT activation and MMP activity [63,66]. In cervical cancer, ZC3H12A binds to XIAP mRNA via its ZF domain and promotes its degradation through its PIN domain RNase activity, thereby inducing apoptosis via the XIAP/caspase/PARP1 pathway [67]. Similarly, in breast cancer, ZC3H12A inactivates IL-17 signaling to suppress tumor growth and metastasis [68] and exerts anti-proliferative effects by inducing G0/G1 cell cycle arrest in triple-negative breast cancer (TNBC) [69].
In gliomas, ZC3H12A promotes angiogenesis through VEGFA-mediated ERK activation, demonstrating pro-tumor activity. Its knockdown suppresses glioma growth and angiogenesis in vivo [70]. Additionally, ZC3H12A also regulates apoptosis by promoting death receptor 5 (DR5) degradation via the autophagy-lysosome pathway, reducing cellular sensitivity to TRAIL- or DR5-mediated apoptosis [71]. Its expression level negatively correlates with sensitivity to DR5-induced apoptosis, highlight its role in promoting tumor immune evasion and survival [71,72]. Recent studies have explored the implications of ZC3H12A-mediated NF-κB inhibition in tumor progression and therapy resistance [73,74].
The dual nature of ZC3H12A highlights its function as a pivotal RNA regulatory hub—its ultimate biological effects are not intrinsic properties but are determined by integrated signaling pathways within specific cellular environments. Its capacity to either suppress or promote tumorigenesis depends on which dominant oncogenic pathways (such as c-Met, Wnt, IL-17, and VEGFA/ERK) are active and subject to its post-transcriptional regulation (Figure 3).
3.2. The “Architect” of the Tumor Immune Microenvironment
Dysfunction of ZC3H12A profoundly reshapes the entire tumor ecosystem. In pancreatic ductal adenocarcinoma (PDAC), IL-1β induces downregulation of ZC3H12A expression, leading to the accumulation of key chemokines and cytokines such as CXCL1, CXCL2, CSF2, and TGFβ. This recruits large numbers of myeloid-derived suppressor cells (MDSCs), which inhibit the antitumor function of cytotoxic T lymphocytes (CTLs) and mediate immune escape in PDAC [75]. Similarly, in gastric cancer (GC), dysregulation of the linc00936/miR-425-3p/ZC3H12A axis similarly suppresses the antitumor immunity of CIK cells and CD4^+^ T cells by promoting the secretion of immunosuppressive factors such as VEGF, IL-10, and TGF-β1, ultimately leading to immune escape and disease progression [76]. These two cases highlight the pivotal role of ZC3H12A in sustaining antitumor immune responses.
In contrast, the tumor-suppressing function of ZC3H12A in colorectal cancer (CRC) exhibits a more complex hierarchy. On one hand, epithelial cell-autonomous mechanisms play a dominant role: ZC3H12A in intestinal epithelial cells negatively regulates the IL-17 signaling pathway by directly degrading NF-κB IZ (IκBζ) mRNA, thereby suppressing gut microbiota-triggered tumor cell proliferation driven by ERK phosphorylation [77]. On the other hand, the tumor microenvironment also actively participates in attacking ZC3H12A: M2 macrophages deliver miR-143-3p to cancer cells via secreted extracellular vesicles. By targeting and inhibiting ZC3H12A, they release its suppression of the transcription factor C/EBPβ, thereby activating pro-cancer signaling and promoting CRC progression [78].
As a pivotal molecular node, the functional state of ZC3H12A directly determines the equilibrium of the tumor ecosystem. Whether driving immune evasion in pancreatic and gastric cancers or mediating cross-talk between epithelial self-defense and microenvironmental signals in colorectal cancer, ZC3H12A inactivation consistently propels more malignant tumor phenotypes. Thus, reshaping the tumor ecosystem via ZC3H12A may emerge as a promising cross-cancer therapeutic strategy.
3.3. The “Game Changer” in CAR-T Immunotherapy
In solid tumor therapy, the key to the success of engineered T cells lies in their persistence—that is, their ability to maintain a population of T cells with stem-like properties and self-renewal capacity (such as TCF-1^+^ TPEX) within the tumor microenvironment characterized by chronic antigen exposure and metabolic stress [79]. Research indicates that knocking out ZC3H12A reprograms CD8^+^ T cells into a unique “long-lived effector” state, significantly enhancing their accumulation, persistent survival, and killing capacity at tumor sites [80]. This process is finely regulated by the transcription factor BATF, which acts as a molecular “variable resistor” to coordinate the balance between effector differentiation and mitochondrial fitness. Notably, BATF deficiency completely reverses the metabolic advantage and persistence phenotype induced by ZC3H12A knockout, revealing the central role of the “ZC3H12A-BATF-mitochondria-OXPHOS” axis in linking T cell functional states to metabolic reprogramming [80]. In CAR-T systems, mechanism studies further revealed that ZC3H12A directly targets Tcf7 mRNA and inhibits TCF-1 protein expression. Its knockout significantly expands the TCF-1^+^ TPEX cell pool, enhances memory-like differentiation and secondary response capacity, thereby markedly improving tumor clearance efficacy and long-term persistence of CAR-T cells in B-ALL models [81].
The most groundbreaking advancement stems from the combined targeting of ZC3H12A and BCOR. Across multiple CAR-T platforms—including CD19, GD2, and others—simultaneous knockout of ZC3H12A and BCOR induces the generation of functionally distinct “tumor-immunological fitness” (TIF) T cells [82]. Mechanistically, ZC3H12A and BCOR jointly regulate a “core stemness program” comprising approximately 216 factors, exhibiting complementary functions: ZC3H12A knockout primarily initiates the stemness program but is accompanied by increased cell death, whereas BCOR knockout preferentially promotes TPEX proliferation and survival. Their synergy is crucial for achieving “both stemness and functionality” under chronic antigenic conditions [83]. Leveraging the unique properties of CARTIF cells, the study also developed GD2-TIF as a platform for single-dose, long-term delivery of biologics. Its demonstrated persistent in vivo persistence and sustained secretion of therapeutic molecules highlight the central role of the ZC3H12A/BCOR axis in coordinating the “stemness-function-metabolism” network and its clinical translational potential [84].
However, safely knocking out ZC3H12A and BCOR has become critical. Traditional CRISPR-Cas systems generate double-strand breaks (DSBs) associated with genomic rearrangements and genotoxicity during multi-site knockouts, inducing programmed cell death [85,86,87]. New research reveals that employing an “orthogonal CRISPR-Cas” strategy enables DSB-free knockout of ZC3H12A and B2M. Simultaneously integrating CAR targeting into the T cell receptor α constant (TRAC) locus significantly reduces the risk of chromosomal translocation (approximately 210-fold), providing a safe and robust manufacturing platform for multi-site engineering [88]. These advances, together with the signals of long-term complete remission observed in preclinical studies, collectively underscore the critical value of “persistence engineering” in overcoming treatment bottlenecks for solid tumors [89].
In solid tumor models treated with humanized engineered T cells, dual knockout of ZC3H12A and Roquin-1 demonstrated stronger antitumor efficacy compared to single knockout. However, some cases developed lymphoproliferative syndrome and related toxicities, suggesting that enhanced therapeutic effects coexist with autoimmune risks [90]. Under these circumstances, ZC3H12A/BCOR dual-targeted CARTIF cells can serve as the core foundation. After thoroughly evaluating the risk-benefit ratio, the combination intervention with Roquin-1 should be prudently considered to balance therapeutic gains against the risk of autoimmune toxicity [82,91]. In vivo CRISPR screening studies confirm that knocking out ZC3H12A or Roquin-1 confers significant expansion advantages to CAR-T cells during early treatment phases. These findings also suggest that the impact of different genetic perturbations on persistence exhibits time- and context-dependent effects, necessitating a balance between therapeutic efficacy and toxicity control [92]. To achieve safe and effective translational application, a transgenic expression system can be designed based on the recognition mechanism of endogenous mRNA 3′ UTR stem-loop structures by ZC3H12A/Roquin-1. This system would be suppressed during quiescence and de-inhibited upon T cell activation, thereby specifically enhancing effector function within the tumor microenvironment (Figure 3) [93].
In summary, by leveraging ZC3H12A/BCOR dual-targeted CARTIF cells as a core platform, prudently integrating enhanced strategies targeting Roquin-1 under clearly defined risk thresholds, and complementing these with precise transgenic regulatory approaches, we anticipate developing next-generation T-cell therapies for tumor treatment that combine sustained efficacy with safety [84,92,93].
4. ZC3H12A-Mediated Systemic Metabolic–Immune Dialog
Nutritional excess and dietary imbalances resulting from modern lifestyles trigger systemic chronic low-grade inflammation through multiple pathways, including lipotoxicity, oxidative stress, and enteric signals. This inflammation subsequently drives metabolic disorders within the pancreas-liver-adipose tissue axis and the onset of related endocrine diseases, forming the key pathological basis of metabolic syndrome [94]. ZC3H12A has been identified as a central node within the intricate network of physiological and pathological processes that make up the “nutrition-inflammation-metabolism” interaction hub.
ZC3H12A expression in subcutaneous adipose tissue exhibits an inverse relationship with BMI. Its suppression in 3T3-L1 adipocytes reduces GLUT4 expression and diminishes glucose uptake, indicating a fundamental role in sustaining adipocyte metabolic competence and insulin sensitivity [95]. In pancreatic β-cells, ZC3H12A fine-tunes stress-responsive transcriptional networks involving FOXO1 and PDX1; impaired insulin secretion upon its downregulation underscores its importance in β-cell functional integrity [96,97].
Clinical observations link serum ZC3H12A levels to diabetic nephropathy progression. Mechanistically, central dapagliflozin administration elevates hypothalamic ZC3H12A, mitigating neuroinflammation and improving renal lipid metabolic reprogramming—supporting its dual role as a biomarker and therapeutic target within the renal-brain axis [98].
Immunometabolically, myeloid-specific ZC3H12A deletion triggers systemic inflammation and perturbs hepatic metabolism, illustrating its homeostatic function across tissues [99]. Consistent with this, reduced ZC3H12A in obese individuals’ mononuclear cells accompanies elevated inflammatory markers, reinforcing its protective role against meta-inflammation [100]. Notably, the ZC3H12A/BCOR-dependent T cell platform permits persistent metabolic modulator delivery, evidenced by GLP-1-secreting GD2-TIF cells thwarting obesity and diabetes in vivo—validating cell-based therapeutic strategies for sustained metabolic-inflammatory rebalancing [84].
In summary, ZC3H12A constitutes a molecular linchpin connecting nutritional excess, metabolic dysfunction, and chronic inflammation. Through its coordinated regulation of adipocyte insulin response, β-cell secretion, and renal metabolic-inflammatory coupling, it articulates core mechanisms of metabolic syndrome. Targeted modulation of ZC3H12A thus offers a compelling strategy for stratified metabolic intervention, spanning lifestyle, hormonal, immunometabolic, and cell-based modalities.
5. New Strategies for Treating Inflammatory and Infectious Diseases with ZC3H12A
5.1. Enhancing ZC3H12A as a Therapeutic Strategy in Inflammatory Diseases
ZC3H12A is a ribonuclease that directly impairs microRNA (miRNA) maturation through cleavage of precursor miRNAs (pre-miRNAs), thereby directly regulating the microRNA maturation process. By antagonizing the Dicer nuclease, it exerts a broad influence on microRNA-mediated gene silencing [101,102,103,104]. In the early stages of inflammation, ZC3H12A regulates inflammatory responses by suppressing the miR-155/RORα axis and preventing excessive NF-κB signaling activation (Figure 4) [105]. Conversely, its downregulation during the late inflammatory phase promotes miR-146a maturation, thereby facilitating inflammation resolution [106,107]. In addition to regulating miRNAs, ZC3H12A can directly degrade the mRNA of various inflammatory mediators, such as IL-6. This has anti-inflammatory and tissue-protective effects in multiple organs, including the cardiovascular system, lungs, liver, kidneys and intestines [108,109,110,111,112,113,114]. In recent years, therapeutic strategies targeting the activation of ZC3H12A have made significant progress. Research indicates that ZC3H12A recognizes stem-loop structures within the 3′ UTR regions of inflammation-related mRNAs, including its own mRNA, and directs their degradation [115]. Based on this mechanism, researchers designed antisense morpholino oligonucleotides (MOs) targeting this autoregulatory site. By specifically blocking ZC3H12A’s degradation of its own mRNA, these MOs elevated its protein expression levels, thereby enhancing its anti-inflammatory function. Crucially, this approach did not impair its ability to recognize other target mRNAs such as IL-6, offering a novel nucleic acid therapeutic strategy for treating inflammatory diseases [116].
5.2. The Dual Role of ZC3H12A in Infectious Diseases
Although the absence of ZC3H12A in infectious diseases may exacerbate autoimmune responses, it may impair host defense in some infections; for example, ZC3H12A knockout enhances type I interferon responses and improves bacterial clearance in Klebsiella pneumoniae (KP) infection [117]. Notably, ZC3H12A directly targets viral RNAs; it degrades spliced HIV-1 transcripts to suppress replication [118,119], cleaves non-ARE regions in coxsackievirus B3 RNA [120], degrades KSHV pre-miRNAs, suppresses Dicer [121], modulates miR-122 biogenesis to inhibit HCV [122], and accelerates HBV RNA decay via recognition of the terminal redundancy region [123]. In chronic viral infections, co-inhibition of BCOR and ZC3H12A promotes stem-like exhausted T cells and improves viral control [83].
Therefore, increasing ZC3H12A activity could be useful in treating inflammatory diseases, while temporarily inhibiting it could help defend against certain infections, demonstrating its context-specific therapeutic potential.
6. Conclusions and Perspectives
In summary, ZC3H12A, as a key regulatory factor with both RNase and deubiquitinating enzyme activities, plays a central role in immune homeostasis, inflammatory responses, and tumorigenesis. In recent years, its breakthrough progress in CAR-T cell therapy has been particularly noteworthy: knocking out ZC3H12A significantly enhances CAR-T cell persistence, stem cell-like properties, and anti-tumor capabilities. Notably, the strategy of co-knocking out ZC3H12A and BCOR successfully induces CAR_TIF_ cells with “tumor immune adaptability,” demonstrating potential for long-term survival and sustained tumor clearance. Despite safety challenges, novel gene editing strategies and precision regulatory systems offer viable pathways for clinical application. Consequently, ZC3H12A has emerged as a key molecular target for optimizing T-cell therapies and advancing tumor immunotherapy.
Moreover, its potential involvement in DNA damage repair remains less explored. DNA damage triggers NF-κB activation, which contributes to cellular response to genotoxic stress. Previous studies indicate that TANK, ZC3H12A, and USP10 can form a deubiquitination complex that suppresses genotoxic NF-κB activation by reducing the ubiquitination levels of TRAF6 and NEMO [29]. This mechanism may help overcome resistance to cancer therapies and alleviate autoimmune pathologies. Recent work from our laboratory identified ZC3H12A as a factor influencing DNA damage repair and radiosensitivity in small cell lung cancer (SCLC) [124]. Future studies should aim to elucidate the specific molecular mechanisms by which ZC3H12A affects DNA damage repair, as well as the precise regulation of ZC3H12A activity and the development of targeted therapeutic agents.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Coussens L.M. Zitvogel L. Palucka A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet?Science 201333928629110.1126/science.123222723329041 PMC 3591506 · doi ↗ · pubmed ↗
- 2Houghton J. Stoicov C. Nomura S. Rogers A.B. Carlson J. Li H. Cai X. Fox J.G. Goldenring J.R. Wang T.C. Gastric cancer originating from bone marrow-derived cells Science 20043061568157110.1126/science.109951315567866 · doi ↗ · pubmed ↗
- 3Mantovani A. Allavena P. Sica A. Balkwill F. Cancer-related inflammation Nature 200845443644410.1038/nature 0720518650914 · doi ↗ · pubmed ↗
- 4Kawai T. Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity Immunity 20113463765010.1016/j.immuni.2011.05.00621616434 · doi ↗ · pubmed ↗
- 5Takeuchi O. Akira S. Pattern recognition receptors and inflammation Cell 201014080582010.1016/j.cell.2010.01.02220303872 · doi ↗ · pubmed ↗
- 6Anderson P. Post-transcriptional control of cytokine production Nat. Immunol.2008935335910.1038/ni 158418349815 · doi ↗ · pubmed ↗
- 7Stoecklin G. Anderson P. Posttranscriptional mechanisms regulating the inflammatory response Adv. Immunol.2006891371668227110.1016/S 0065-2776(05)89001-7 · doi ↗ · pubmed ↗
- 8Katsanou V. Dimitriou M. Kontoyiannis D.L. Post-Transcriptional Regulators in Inflammation: Exploring New Avenues in Biological Therapeutics Ernst Schering Foundation Symposium Proceedings 2006375710.1007/2789_2007_03817824180 · doi ↗ · pubmed ↗