Hypertension Resistant to RAAS Inhibitors as a Prognostic Indicator for Rapid Progression to ESRD in ADPKD: A Ten-Year Follow-Up
Andrea Angioi, Doloretta Piras, Nicola Lepori, Matteo Floris, Gianfranca Cabiddu, Antonello Pani

TL;DR
Hypertension that doesn't respond to RAAS inhibitors predicts faster kidney disease progression in ADPKD patients.
Contribution
Identifies RAAS-resistant hypertension as a novel independent predictor of rapid ESRD progression in ADPKD.
Findings
RAAS-resistant hypertension is associated with a 27% higher odds of rapid eGFR decline in ADPKD patients.
Resistant hypertension may indicate a more aggressive cystic phenotype and intrarenal dysregulation.
Early identification of resistant hypertension could improve risk stratification and treatment decisions.
Abstract
Background: Autosomal dominant polycystic kidney disease (ADPKD) is characterized by progressive renal cyst development and variable trajectories toward end-stage renal disease (ESRD). Hypertension is both common and prognostically significant in ADPKD. However, the escalating need for antihypertensive agents beyond RAAS inhibition on disease progression remains underexplored. Methods: We conducted a retrospective, single-center cohort study including 133 ADPKD patients followed for a median of 5 years. Baseline clinical, biochemical, and genetic data were collected. The primary outcome was a ≥25% decline in eGFR over 5 years. All patients achieved a blood pressure target range of 110/70 to 130/85 mmHg during follow-up. Univariate and multivariate logistic regression analyses were performed to identify predictors of rapid progression. Results: Patients with hypertension resistant to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
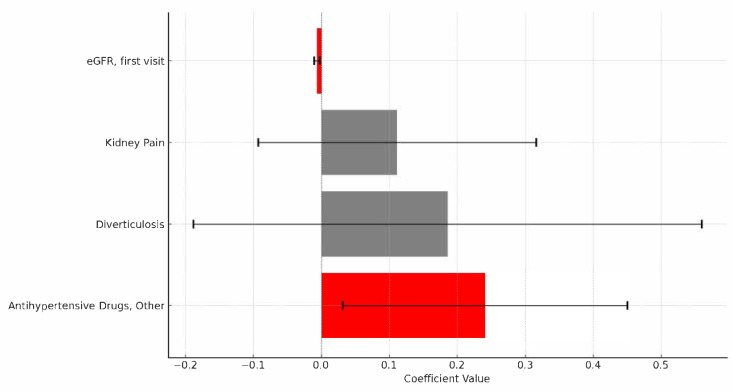 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Kidney Cyst Diseases · Biomedical Research and Pathophysiology · Renal Diseases and Glomerulopathies
1. Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a major health concern because it is the most common hereditary kidney disease worldwide, affecting about 12.5 million individuals [1]. Characterized by multiple cysts mainly in the kidneys and liver, ADPKD presents a significant healthcare challenge because it can progress to kidney failure, ultimately needing replacement therapy.
Approximately 85% of ADPKD cases result from mutations in the PKD1 gene, while mutations in the PKD2 gene account for about 10–15% of cases [2]. Mutations in genes involved in protein biogenesis in the endoplasmic reticulum, such as ALG9, GANAB, SEC63, SEC61B, DNAJB11, and ALG8, can also cause cystic kidney and liver diseases by disrupting the production or function of polycystin-1 [3]. These proteins are vital components of primary cilia in tubular epithelial cells, where they regulate cell growth, programmed cell death, water and electrolyte influxes and effluxes, and extracellular matrix remodeling [3]. The genetic defect initially causes the formation of cystic dilation (diffuse), and then, in less than 5% of tubules, the development of cysts that vary from microscopic to large enough to be visible on ultrasound or RMN. These cysts gradually enlarge, impairing kidney function through both mechanical pressure and functional disruption.
Despite this genetic burden, the progression of ADPKD varies greatly among individuals, with some developing end-stage renal disease (ESRD) early in life. Others, however, retain moderate kidney function into old age, with the same mutation. This variability highlights a significant gap in our understanding of ADPKD’s natural course and the epigenetics that influence disease progression [4].
In the literature, prognostic scores are used to predict, based on clinical and genetic criteria, whether an individual will have a rapid or slow progression to ESRD. The most commonly used score in clinical practice is the PRO-PKD score. This prognostic model utilizes genetic and clinical data to identify key factors, including gender, hypertension before age 35, the first urologic event before age 35, PKD2 mutation, and non-truncating or truncating PKD1 mutation. These factors enable the stratification of patients into low, intermediate, and high-risk categories, each associated with different median ages for ESRD onset [5]. Other models have been described in the literature that consider additional variables (e.g., early-onset hypertension, height-adjusted mean kidney length); notably, every score highlights the degree of genetic mutation as a critical independent predictor [6,7]. However, in real-world clinical settings, the PRO-PKD score identifies fewer rapid progressors compared to the Mayo score, and it is not universally effective across all cohorts [8].
Uncovering the factors that influence disease progression in ADPKD can provide valuable insights for customizing individual treatment strategies. Therefore, this study aimed to examine how specific clinical features and baseline biochemical parameters affect CKD progression in ADPKD patients within our single-center cohort over a 10-year follow-up period.
2. Materials and Methods
2.1. Study Design and Participants
This retrospective, a single-center cohort study was conducted at a tertiary care institution (ARNAS “G. Brotzu,” Cagliari, Italy), involving patients in a dedicated outpatient clinic specialized in Polycystic Kidney Disease and other Cystic Disorders with 172 patients on follow-up on a regular basis.
Inclusion criteria: (1) in cases of a positive family history, a diagnosis of ADPKD was based on Pei’s ultrasonographic criteria: age 15–39 years, ≥three renal cysts (unilateral or bilateral); age 40–59 years, ≥2 cysts in each kidney; age ≥ 60 years, ≥four cysts in each kidney [9]. (2) In case of a negative family history: presence of 10 or more cysts (≥5 mm) in each kidney, especially if the kidneys are enlarged or liver cysts are observed, and when there are no apparent signs of a different cystic disorder. (3) Genetic diagnosis by identifying pathogenic mutations in the PKD1 or PKD2 genes.
Exclusion criteria included (1) patients classified as atypical ADPKD (Class 2 Mayo ADPKD classification); (2) less than two documented visits; (3) insufficient medical record data for essential outcome analysis; (4) patients younger than 18 years.
Of 172 patients diagnosed and managed at our institution between January 2018 and December 2024, 133 were eligible.
2.2. Data Collection
Demographic, clinical, genetic, therapeutic, and biochemical data were retrospectively obtained from electronic medical records (Table 1 and Table 2). Collected baseline characteristics included age, sex, body mass index (BMI), body surface area (BSA), gene mutation (PKD1, PKD2, or unidentified mutations), extrarenal manifestations, family history, baseline kidney volume (MRI-derived), baseline kidney function parameters (serum creatinine, eGFR calculated by CKD-EPI 2021 equation, creatinine clearance), 24 h proteinuria, hypertension, antihypertensive treatments (RAAS inhibitors including ACE inhibitors or ARBs, and other antihypertensive agents), and tolvaptan. The PRO-PKD score was calculated based on genetic and clinical factors (gender, hypertension before age 35 years, urologic complications before age 35 years, PKD mutation type, and mutation characteristics). Kidney pain was defined as recurrent flank pain explicitly attributed to renal cyst enlargement or complications, documented clinically, and occurring more than twice per year.
2.3. Outcomes and Follow-Up
The primary outcome measured was the decline in kidney function, specifically the predicted percentage reduction in eGFR at five years, with a clinically significant threshold set at a 25% reduction [10]. The annualized eGFR slope (mL/min/1.73 m^2^ per year) was calculated for each patient to evaluate the course of kidney function decline over the follow-up period. The median follow-up duration was five years, with routine clinical and biochemical assessments conducted every 3–12 months as needed. All patients achieved guideline-concordant BP targets: ≤110/75 mmHg by HBPM in adults 18–49 with CKD G1–G2, and standardized office SBP < 120 mmHg in adults ≥50 years or with lower eGFR, as per KDIGO 2025 (ADPKD) [11]. The target was reached and monitored by a daily blood pressure control. Patients were trained by their general practitioner to follow this HBPM protocol (cuff sizing/positioning, posture and 5 min rest, morning/evening measurements, and diary recording); technique and adherence were reviewed at routine visits.
2.4. Statistical Analysis
Continuous variables were described as medians with interquartile ranges (IQRs) due to their non-normal distribution, as confirmed by the Shapiro–Wilk test, or as means ± standard deviations (SDs) when variables exhibited a normal distribution. Categorical variables were presented as frequencies and percentages. Differences in baseline characteristics before and after propensity score matching were evaluated using Student’s t-test for normally distributed variables and the Mann–Whitney U test for skewed variables, while categorical variables were compared with Chi-square or Fisher’s exact tests, as appropriate.
Univariate logistic regression analysis identified clinical and biochemical factors associated with the outocome, expressed as odds ratios (OR) with 95% CI.
All variables meeting this threshold were then simultaneously entered into a multivariate logistic regression model using the Enter (forced-entry) method in JASP (Version 0.19.3). This approach ensured that the effect of each covariate was estimated while adjusting for all others, without resorting to stepwise selection.
2.5. Software and Ethics
All statistical analyses were conducted using JASP software, version 0.19.3 (JASP Team, Amsterdam, The Netherlands). The retrospective study was approved by the Institutional Ethics Committee of the Azienda Ospedaliero-Universitaria di Cagliari (protocol number PG/2019/16560, approved on 27 November 2019). Informed consent was obtained through the protocol guidelines.
3. Results
3.1. Patient Demographics and Clinical Characteristics
The study included 133 patients diagnosed with ADPKD, predominantly harboring PKD1 mutations (73.68%), followed by PKD2 mutations (10.53%) and unidentified mutations (15.79%). Males comprised 44.36% of the cohort, and 85.00% reported a positive family history. Frequent extrarenal manifestations included cardiac hypertrophy (37.59%), intraductal papillary mucinous neoplasms (IPMN, 17.29%), and nephrolithiasis (8.27%). Hypertension was prevalent in 55.63%, and frequent kidney pain (>2 episodes/year) was reported by 52.63% (Table 1 and Table 2).
3.2. Treatment and Therapeutic Patterns
ACE inhibitors were the most commonly prescribed antihypertensive therapy, used by 64 patients (48.12%), while ARBs were given to 23 patients (17.29%). A combination of RAAS inhibitors (either ACE inhibitors or ARBs) plus additional antihypertensive medications was needed for 37 patients (27.8%). In total, 50 patients (37.6%) received only RAAS inhibitors without extra antihypertensive agents. A smaller group (7.5%, 10 patients) was managed solely with antihypertensive medications that did not include RAAS inhibitors. Tolvaptan was administered to 17 eligible patients (12.8%), whereas 36 patients (27.1%) did not receive any antihypertensive therapy due to hypotension.
3.3. Biochemical Parameters and eGFR Progression
The baseline median eGFR was 67.9 mL/min/1.73 m^2^ (IQR 31.0), significantly declining to 48.9 mL/min/1.73 m^2^ (IQR 32.9) at five years, reflecting a median decrease of 19.0 mL/min/1.73 m^2^ (IQR 11.89). The annual median eGFR decline was 3.8 mL/min/1.73 m^2^ per year. The baseline kidney volume, assessed by MRI, was 1379.95 mL (IQR, 881.8).
3.4. Identification of Predictive Factors for CKD Progression
Univariate logistic regression identified significant predictors of a 25% eGFR reduction at 5 years (Table S1). Use of RAAS inhibitors plus antihypertensive drugs was strongly associated with the progression of kidney disease (OR 3.03, 95% CI 1.16–7.914, p = 0.02360), as was average creatinine at first visit (OR 3.453, 95% CI 1.274–9.362, p = 0.01489). Initial eGFR (OR 0.969, 95% CI 0.952–0.987, p = 0.00059) and creatinine clearance (OR 0.971, 95% CI 0.955–0.987, p = 0.00037) showed protective effects. ACE inhibitors (OR 1.475, p = 0.39346) and ARBs (OR 1.247, p = 0.69460) alone were not significantly associated with progression (Table 3).
Multivariate linear regression refined these findings (Table 4). A lower initial eGFR was a significant predictor of greater eGFR decline (OR 0.993, CI 95% 0.989–0.997; p = 0.0012), as was the use of other antihypertensive drugs (OR 1.272, CI 95% 1.032–1.569; p = 0.0248). Diverticulosis and kidney pain were included in the multivariate analysis since they were close to the significance threshold in the univariate analysis but did not reach statistical significance in the multivariate model (p > 0.05). This approach does not account for time-to-event information, which is acknowledged as a limitation compared with Cox proportional-hazards modeling.
4. Discussion
In this single-center cohort of ADPKD patients, we found that hypertension resistant to RAAS, operationally defined as the need for additional antihypertensive therapy beyond RAAS inhibition, is a significant marker of rapid disease progression. Patients who required more than a single RAAS blocker to control blood pressure had higher odds of experiencing a ≥25% decline in eGFR over 5 years. On multivariate analysis adjusting for key baseline variables (including age, sex, and baseline eGFR), hypertension resistant to RAAS remained an independent predictor of kidney function decline (adjusted OR 1.27, 95% CI 1.03–1.57; p = 0.0248) (Figure 1). These findings suggest that difficulty in achieving blood pressure control, despite optimal first-line therapy, is not merely a coincidental comorbidity but rather a proxy for a more aggressive cystic disease. This is biologically plausible: extensive cyst burden in ADPKD activates the intrarenal RAAS, leading to early and severe hypertension. Indeed, the severity of hypertension correlates with TKV and cyst growth rate in ADPKD. In clinical terms, hypertension resistant to RAAS serves as a red flag for clinicians, identifying patients with ADPKD who are on a trajectory of rapid renal function loss [12,13].
Our results align with and extend prior observations regarding hypertension and ADPKD progression. It has long been recognized that hypertension is highly prevalent in ADPKD and portends worse outcomes [14]. Approximately 50–70% of ADPKD patients develop hypertension before any significant loss of GFR, often decades earlier than in hypertensive patients without ADPKD. Early-onset hypertension in ADPKD is strongly associated with faster cyst growth and earlier onset of end-stage renal disease (ESRD). Previous longitudinal studies have identified baseline hypertension as an independent risk factor for kidney function decline and progression to ESRD in ADPKD [15,16]. Notably, the PROPKD score incorporates “early hypertension” (onset before age 35) as a key risk factor, along with gene mutation type and urologic complications [5].
Our findings build upon this knowledge by focusing on the subset of patients with hypertension to RAAS. While prior studies emphasized the presence or absence of hypertension, we show that the degree of blood pressure control (or lack thereof) on standard therapy carries additional prognostic significance. In other words, it is not only whether an ADPKD patient is hypertensive, but how difficult that hypertension is to treat, that predicts renal outcome. This nuance has not been explicitly addressed in earlier ADPKD cohorts and provides a new perspective on risk stratification.
The results of our study are also consistent with broader observations in chronic kidney disease (CKD) populations. In non-ADPKD CKD, resistant or poorly controlled hypertension has been linked to accelerated renal function loss and higher rates of reaching ESRD [15].
The clinical implications of these findings are immediate and practical. First and foremost, hypertension resistant to RAAS, if confirmed in other cohorts, should be recognized as a high-risk feature in ADPKD. In routine practice, it is relatively easy to identify patients who meet this criterion (those already on an ACE inhibitor or ARB who require one or more additional drugs) to achieve blood pressure targets. Such patients should be flagged for enhanced risk stratification and closer follow-up, prompting clinicians to actively evaluate for rapid disease progression. This could include obtaining baseline and follow-up measurements of TKV (e.g., via MRI) sooner, to see if the patient falls into a high-risk Mayo class. It could also include considering genetic testing if not already done, since a patient with difficult-to-control hypertension may have an underlying high-impact PKD1 mutation or other genetic modifiers of severity. By incorporating hypertension resistant to RAAS into the risk assessment, clinicians can identify “rapid progressors” earlier in the disease course. This aligns closely with KDIGO 2025 guidance, which stresses early identification of patients likely to benefit from therapy to delay progression [11]. Another significant implication is the potential to initiate disease-modifying treatments earlier in patients with hypertension resistant to RAAS. The vasopressin V2-receptor antagonist tolvaptan is currently the only approved therapy shown to slow ADPKD progression. Tolvaptan has been demonstrated in large trials to slow the rate of increase in TKV and the decline in GFR in ADPKD [16,17].
However, we acknowledge several significant limitations of our cohort. (1) The study’s retrospective observational design inherently limits causal inferences. (2) The lack of comprehensive genetic data (15.79% of patients have an unknown mutation). (3) It was conducted at a single tertiary care center, limiting generalizability. (4) Our criterion of “beyond RAAS inhibition” captures a slightly broader group, potentially including some patients who needed just a second agent. (5) Non-adherence may have played a role. (6) As with any observational study, there may be residual confounders. (7) The outcome we used, a 25% decline in eGFR, while clinically meaningful, is still a surrogate endpoint of “hard” endpoints like ESRD, but our cohort’s follow-up (median 5.2 years) and sample size yield relatively few of those events to power the analysis.
5. Conclusions
In this single-center cohort of ADPKD patients, hypertension resistant to RAAS, defined as the need for additional agents beyond RAAS inhibition, emerged as an independent predictor of accelerated eGFR decline over 5 years. Its presence may reflect a more aggressive cystic burden and intrarenal hemodynamic dysregulation. Early identification of hypertension resistant to RAAS could prompt intensified monitoring and earlier initiation of disease-modifying interventions. Validation in larger cohorts is warranted.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Spithoven E.M. Kramer A. Meijer E. Orskov B. Wanner C. Abad J.M. ArestéN. Alonso de la Torre R. Caskey F. Couchoud C. Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: Prevalence and survival—An analysis of data from the ERA-EDTA Registry Nephrol. Dial. Transplant.201429 iv 15iv 2510.1093/ndt/gfu 01725165182 PMC 7611099 · doi ↗ · pubmed ↗
- 2Hoefele J. Mayer K. Scholz M. Klein H.-G. Novel PKD 1 and PKD 2 mutations in autosomal dominant polycystic kidney disease (ADPKD)Nephrol. Dial. Transplant.201126218121882111567010.1093/ndt/gfq 720 · doi ↗ · pubmed ↗
- 3Lanktree M.B. Haghighi A. di Bari I. Song X. Pei Y. Insights into Autosomal Dominant Polycystic Kidney Disease from Genetic Studies Clin. J. Am. Soc. Nephrol.20211679010.2215/cjn.0232022032690722 PMC 8259493 · doi ↗ · pubmed ↗
- 4Agborbesong E. Li L.X. Li L. Li X. Molecular Mechanisms of Epigenetic Regulation, Inflammation, and Cell Death in ADPKD Front. Mol. Biosci.2022992242810.3389/fmolb.2022.92242835847973 PMC 9277309 · doi ↗ · pubmed ↗
- 5Cornec-Le Gall E. Audrézet M.P. Rousseau A. Hourmant M. Renaudineau E. Charasse C. Morin M.P. Moal M.C. Dantal J. Wehbe B. The PROPKD Score: A New Algorithm to Predict Renal Survival in Autosomal Dominant Polycystic Kidney Disease J. Am. Soc. Nephrol. JASN 2016279429512615060510.1681/ASN.2015010016 PMC 4769200 · doi ↗ · pubmed ↗
- 6Chen E.W. Chong J. Valluru M.K. Durkie M. Simms R.J. Harris P.C. Ong A.C. Combining genotype with height-adjusted kidney length predicts rapid progression of ADPKD Nephrol. Dial. Transplant.20243995696610.1093/ndt/gfad 27038224954 · doi ↗ · pubmed ↗
- 7Wigerinck S. Schellekens P. Smith B.H. Hanna C. Dachy A. Chedid M. Borghol A.H. Senum S.R. Bockenhauer D. Harris P.C. Characteristics of patients with autosomal polycystic kidney disease reaching kidney failure by age 40Pediatr. Nephrol.2025401997200710.1007/s 00467-024-06652-739891678 PMC 12336910 · doi ↗ · pubmed ↗
- 8Capelli I. Aiello V. Carretta E. Graziano C. Sciascia N. Corsi C. Mantovani V. Monteduro F. Seri M. La Manna G. P 0085 MAYO and PRO-PKD score concordance for progression of renal failure evaluation in ADPKD patients Nephrol. Dial. Transplant.202035 gfaa 142.P 008510.1093/ndt/gfaa 142.P 0085 · doi ↗