Investigation of the Relevance of CYP3A4 Inhibition on the Pharmacokinetics of the Novel P2X3 Antagonist Filapixant: Results of In Vitro Explorations and a Fixed-Sequence Clinical Trial with Itraconazole in Healthy Volunteers
Klaus Francke, Antje Rottmann, Stefan Klein, Joachim Höchel, Christian Friedrich

TL;DR
This study examines how CYP3A4 inhibition affects the drug levels of filapixant, a new treatment for chronic cough, in lab and clinical trials.
Contribution
The study provides new insights into the CYP3A4-mediated metabolism of filapixant and its clinical implications.
Findings
Filapixant is primarily metabolized by CYP3A4 with moderate to high turnover in human liver microsomes.
Itraconazole significantly increased filapixant's AUC and Cmax, showing it is a moderately sensitive CYP3A4 substrate.
Co-administration of itraconazole prolonged filapixant's half-life and affected its bioavailability and elimination.
Abstract
Hypersensitized P2X3 receptor signaling has been described to play a role in several disorders, including chronic cough. The goal of our in vitro and in vivo studies was to investigate the biotransformation and the influence of CYP3A4 inhibition on the pharmacokinetics of the selective P2X3 antagonist filapixant. Metabolic turnover of filapixant in human liver microsomes and hepatocytes was moderate to high, indicating a complex metabolic pattern with mainly oxidative biotransformation. In recombinant CYP enzymes, depletion of filapixant was observed mainly with CYP3A4 and, to a significantly lesser extent, with CYP1A1, 2D6, 2J2, and 3A5. Drug depletion of [3H]filapixant and metabolite formation in human liver microsomes was significantly inhibited in the presence of strong CYP3A4 inhibitors, whereas other CYP isoform–selective inhibitors showed no or very minor effects.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
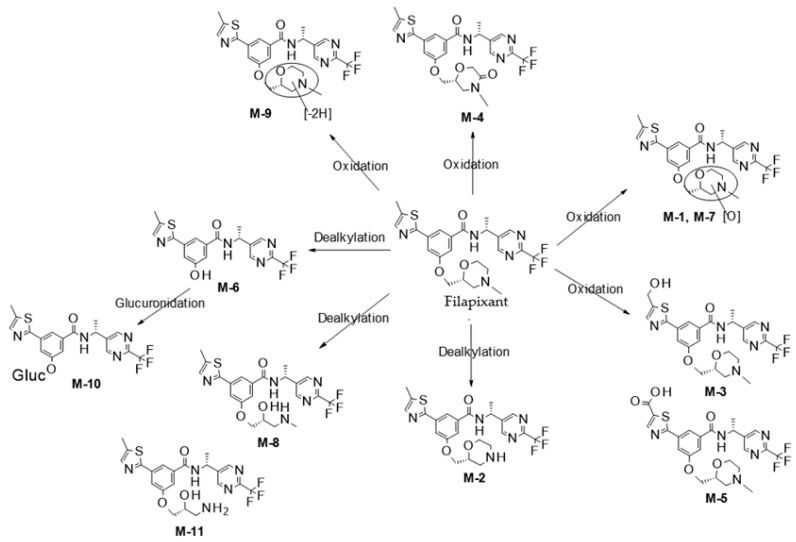 Figure 1
Figure 1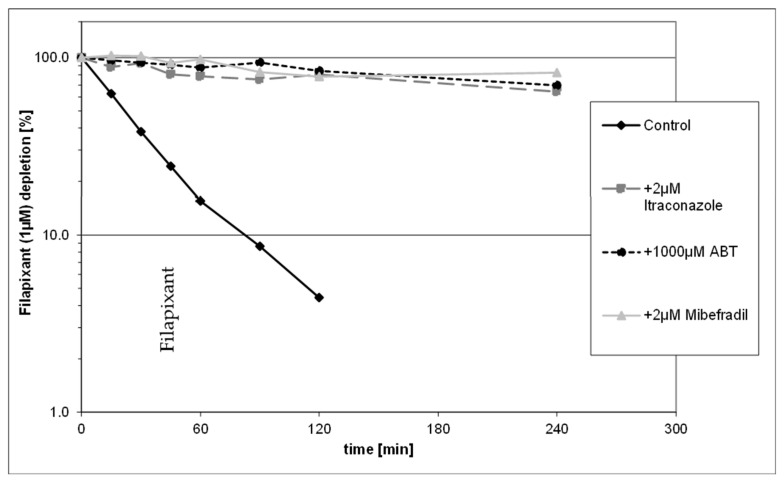 Figure 2
Figure 2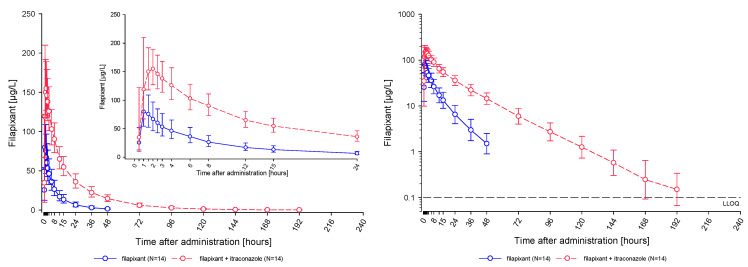 Figure 3
Figure 3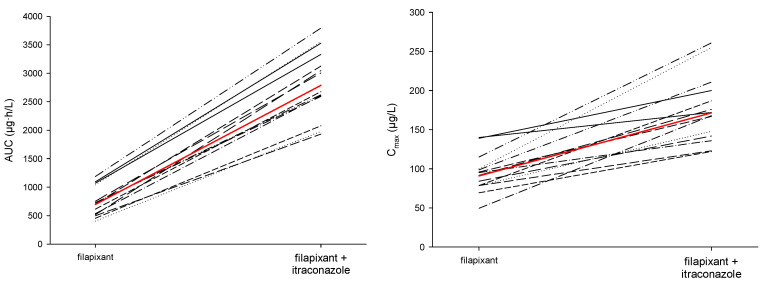 Figure 4
Figure 4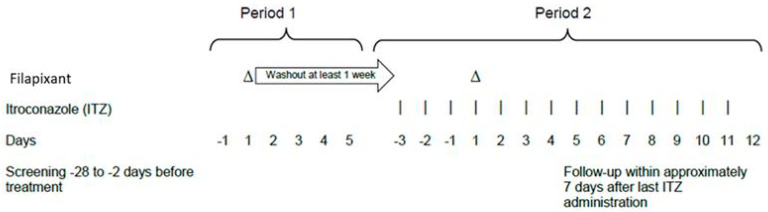 Figure 5
Figure 5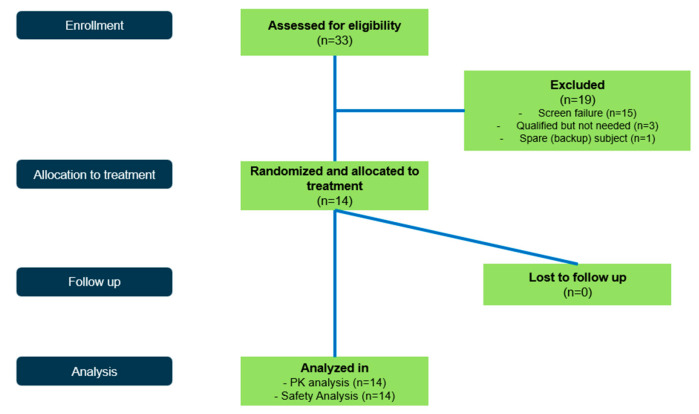 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRespiratory and Cough-Related Research · Gastroesophageal reflux and treatments · Asthma and respiratory diseases
1. Introduction
Several P2X3 receptor antagonists have been in development for the management of refractory chronic cough and have also been tested for other conditions such as endometriosis, osteoarthritis, and bladder disorders. The first clinically investigated P2X3 antagonist, gefapixant, reported efficacy in chronic cough, osteoarthritis, and bladder pain syndrome, which was hampered by a high frequency of taste disturbances. These side effects were considered likely related to the blockade of the P2X2/3 receptor heteromer in the taste-sensing system. Therefore, P2X3-selective receptor antagonists have been developed with the hope to avoid those findings. This has been only partially achieved—the frequency of taste-related side effects with the selective P2X3 antagonist eliapixant was reported to be considerably reduced compared to gefapixant; however, taste disturbances were still seen in a relevant number of patients [1]. Interestingly, another newly developed and even more selective P2X3 antagonist, filapixant (BAY 1902607), showed a taste disturbance frequency being substantially higher than for eliapixant, indicating that other factors besides selectivity such as pharmacokinetic (PK) properties might play a role. Filapixant is a P2X3 antagonist with approximately 100-fold selectivity over the P2X2/3 heteromeric receptor developed for the indication of refractory chronic cough. It has been characterized in initial phase I and phase IIa studies, where it was overall safe and well tolerated and showed its potential in treating patients with refractory chronic cough [2,3]. In phase I studies, filapixant was rapidly absorbed, with maximum concentrations reached about 1–2 h after administration and showed dose-proportional and time-linear PK. The observed terminal half-life of ~6–10 h makes it suitable for once-daily treatment. Co-administration of filapixant had no relevant effect on the PK of midazolam in humans [3].
In this paper, we report the results of in vitro investigations on the metabolism of filapixant and the results of a subsequent clinical drug–drug interaction study with filapixant and itraconazole, a potent cytochrome P450 3A4 (CYP3A4) and P-glycoprotein (P-gp) inhibitor. The human study evaluated the drug–drug interaction potential with the strong CYP3A4/P-gp inhibitor itraconazole to allow dosing recommendations for comedications of filapixant in future clinical studies. Itraconazole was chosen as prototypical inhibitor according to relevant guidelines [4,5].
2. Results
2.1. In Vitro Metabolite Profiling and Enzymes Involved in Metabolism of Filapixant
The metabolic turnover of filapixant in human liver microsomes and hepatocytes was moderate to high showing a complex metabolic pattern (see Appendix C). Mainly oxidative biotransformation at the methyl morpholine moiety was observed (see Figure 1). Oxidation/hydroxylation forming M 1, M 4, and M 7 as well as dealkylation to M 2, M 6, and M 8 were the major primary metabolic pathways. Oxidation at the methyl thiazole moiety forming M 3 and subsequent oxidation to carboxylic acid M 5 was observed to a minor extent. In human hepatocytes, O dealkylation product M 6 is completely glucuronidated to M 10.
In recombinant cytochrome P450 (CYP) enzymes, relevant depletion of filapixant was observed, mainly applying CYP3A4 and to a substantially lesser extent also with CYP1A1, 2D6, 2J2, and 3A5 (Table 1). The apparent intrinsic clearance (CL_int_) of filapixant at concentrations of 0.1 and 1 μM was determined with CYP isoforms (1A1, 2D6, 2J2, and 3A4), thus confirming the dominant role of CYP3A4, with CL_int_ values at least 10-fold higher relative to the other CYP isoforms tested. Determination of CLint of filapixant in recombinant CYP1A1, CYP2D6, CYP2J2, and CYP3A4 resulted in the following CLint values: 0.217, 0.0618, and 0.126 and 1.32 µL/min/pmol CYP, respectively (for 1 µM). Incubation of 0.1 µM filapixant in the presence of CYP1A1, CYP2D6, and CYP2J2 resulted in comparable CLint values: 0.252, 0.0716, and 0.132 and 1.66 µL/min/pmol CYP, respectively.
Drug depletion of [^3^H]filapixant and the formation of metabolites in human liver microsomes was inhibited in the presence of strong CYP3A4 inhibitors (itraconazole, mibefradil, and azamulin), while other CYP isoform–selective inhibitors showed no or very minor inhibitory effects on filapixant biotransformation.
Incubation of 1 µM filapixant in human hepatocytes in presence and absence of CYP isoform selective inhibitors underlined the results from previous investigations. In the presence of strong CYP3A4 inhibitors (mibefradil and itraconazole) as well as the pan-CYP inhibitor aminobenzotriazole (ABT), the apparent CL_int_ of filapixant was reduced by about 90 to 95% compared to untreated control, thus confirming the dominant role of CYP3A4 (Figure 2). The concentration of 2 µM itraconazole in the in vitro assay is in the range of the itraconazole plasma concentration in the clinical DDI study (see Section 2.2.1)
2.2. Clinical Study
Overall, 33 participants were screened. Of these, 19 participants were screening failures, and 14 participants were assigned to treatment and completed the study (Figure A1).
Participants were white males with a mean age (±standard deviation, SD) of 47.1 ± 6.2 years, and a mean body mass index (±SD) of 24.5 ± 2.1 kg/m^2^.
2.2.1. Pharmacokinetic Results
A single oral dose of 80 mg filapixant was rapidly absorbed under fasted conditions. Median time to reach maximum observed plasma concentrations of filapixant was approximately 1 h, without as well as with, itraconazole co-administration (Figure 3).
Co-administration of itraconazole markedly increased the systemic exposure of filapixant: The observed mean peak plasma concentrations (C_max_) were nearly twice as high after co-administration of itraconazole than after administration without itraconazole (172 µg/L and 90.8 µg/L, respectively). The observed mean exposure (AUC) was increased to a 4-times higher exposure (2790 µg·h/L vs. 695 µg·h/L; Table 2). As displayed in Figure 4, increases in exposure (AUC and C_max_) were consistent across participants.
Geometric mean terminal half-life (t_1/2_) of filapixant after single-dose administration was longer when filapixant was given together with itraconazole (22.8 h) compared to filapixant alone (12.1 h).
The point estimates (LS-means) and 2-sided 90% confidence intervals (CIs) of the main PK metrics AUC and C_max_ for filapixant resulting from an analysis of variance model are given in Table 3. Co-administration of the strong CYP3A4/P-gp inhibitor itraconazole markedly and significantly increased the systemic exposure of filapixant resulting in 1.89-fold higher mean C_max_ (90% CI: 1.67; 2.15) and 4.01-fold higher mean exposure (AUC, 90% CI: 3.66; 4.39).
Mean maximum concentrations of itraconazole were observed on study day 4 and were 1014.6 µg/L, indicating the achievement of adequate itraconazole exposure for CYP3A4/P-gp inhibition.
2.2.2. Safety Results
In total, seven of the fourteen participants experienced treatment emergent adverse events (TEAEs). There was no apparent difference in the frequency, intensity, and character of the TEAEs between the two treatment periods of the study. The most frequently reported TEAE assessed as related to filapixant was headache (two participants). None of the participants reported any taste-related adverse event.
Overall, there were no clinically relevant trends in the safety laboratory parameters of the participating participants after the administration of filapixant with and without itraconazole. There were no clinically relevant findings observed in blood pressure, pulse rate measurements, and the electrocardiogram assessments.
3. Discussion
The overall aim of our investigations was to evaluate the drug–drug interaction potential of the strong CYP3A4/P-gp inhibitor itraconazole on the PK of orally administered filapixant to allow recommendations whether and how to use filapixant in the presence of CYP3A4 inhibitors in future studies and potentially in medical practice.
Incubations of 1 µM [^3^H]filapixant were performed in human liver microsomes and human hepatocytes in the presence or absence of CYP isoform–selective inhibitors. Drug depletion and formation of metabolites were inhibited in the presence of strong CYP3A4 inhibitors, while other CYP isoform–selective inhibitors showed no or very minor inhibitory effects on [^3^H]filapixant biotransformation. In the presence of strong CYP3A4 inhibitors, the apparent CL_int_ of filapixant was reduced by about 90 to 95% compared to untreated control. Thus, it was concluded that metabolization by CYP3A4 is the main elimination pathway for filapixant indicating a substantial risk for exposure increases in filapixant in the presence of CYP3A4 inhibitors in a clinical setting, and thus a clinical drug–drug interaction study was subsequently conducted.
Of note, filapixant shows in vitro a concentration dependent moderate to high permeability with a saturable efflux. This efflux is mediated by P-gp, thus filapixant is a P-gp substrate. Due to the saturable efflux at higher concentration and the moderate to high permeability, the overall effect of P-gp on the absorption of filapixant is considered to be of limited importance (Bayer AG; data on file).
For the clinical study and in accordance with relevant guidelines [4,5], itraconazole was chosen as a strong index CYP3A4/P-gp inhibitor to characterize a worst-case scenario. Itraconazole was given over 14 days with a single dose of filapixant co-administered one hour before the fourth itraconazole dose. Three days pretreatment were chosen to allow itraconazole to reach plasma concentrations leading to maximum CYP3A4 and P-gp inhibition before concomitant administration of filapixant. After the concomitant administration on day 4, itraconazole treatment was continued for another nine days to maintain maximum CYP3A4/P-gp inhibition during the whole elimination phase of filapixant, which was expected to be prolonged due to itraconazole related inhibition of the main metabolic pathway. The concomitant dosing of filapixant on day 4 was performed 1 h after itraconazole to guarantee maximum itraconazole concentrations and inhibition of CYP3A4 and P-gp at the gut wall during filapixant absorption.
The observed PK data demonstrated that a single oral dose of 80 mg filapixant was rapidly absorbed. The concomitant administration of itraconazole affected C_max_ of filapixant, but not t_max_. Median t_max_ of filapixant was about 1 h in both study periods. However, the range of individual t_max_ values was greater in the co-administration period, indicating an inter-individually variable effect of itraconazole on the absorption of filapixant.
Co-administration of the strong CYP3A4/P-gp inhibitor itraconazole markedly and significantly increased the systemic exposure of filapixant in all treated participants resulting in 1.89-fold mean peak concentrations (C_max_, 90% CI: 1.67; 2.15) and 4.01-fold mean exposure (AUC, 90% CI: 3.66; 4.39) compared to administration of filapixant alone. This effect confirmed the assumptions from prior in vitro studies with filapixant.
Geometric mean terminal half-life (t_1/2_) of filapixant after single-dose administration was—as expected—substantially longer in when participants received filapixant with itraconazole (22.8 h) than after administration of filapixant alone (12.1 h).
In summary, the changes in both C_max_ and half-life together with the overall change in AUC indicate that both clearance and bioavailability were affected by itraconazole and confirm predictions based on in vitro data. Considering the saturable efflux of filapixant due to P-gp dependent transport and the overall moderate to high permeability of the compound, the observed effect is primarily attributed to CYP3A4, while a minor contribution of P-gp cannot be excluded.
Observed maximum plasma concentrations of itraconazole on day 4 were similar to previously observed results and indicate adequate inhibition of CYP3A4 and P-gp [6,7].
Filapixant was well tolerated after a single oral dosing of 80 mg filapixant and in combination with multiple daily oral doses of 200 mg itraconazole. All TEAEs were of mild intensity. There was no apparent difference in the frequency of TEAEs with filapixant alone or combined with itraconazole, including taste-related adverse events. No relevant changes in vital signs, electrocardiograms, or laboratory values have been observed. The absence of taste-related adverse events is consistent with the previously reported multiple-dose escalation study, where taste-related adverse events were prominently seen with a dose leading to maximum concentrations above 400 µg/L, which is about 2-fold higher than the maximum concentrations reached when filapixant 80 mg was co-administered with itraconazole. Thus, it can be assumed that the threshold for taste-related adverse events with filapixant is somewhere between 200 and 400 µg/L.
Based on the observed magnitude of the interaction, a filapixant dose adaptation or exclusion of strong CYP3A4/P-gp inhibitors (at least for long term co-administration) is suggested if the compound is progressed into further development. Otherwise, the risk of taste-related side effects is expected to substantially increase as filapixant plasma concentrations will be reached that have been shown previously to lead to a high frequency of taste-related side effects.
Limitations of the study are the relatively small sample size, the single dose treatment of filapixant, the fixed sequence of treatments, and that only healthy young male individuals were included in the study for safety reasons. Those limitations were considered acceptable for the following reasons. The limited sample size is offset by the well-controlled study conditions that allow to reduce variability and thus to obtain an accurate estimate of the interaction magnitude suitable to make recommendations for subsequent studies. A single dose of filapixant was considered meaningful, considering the dose proportional and time linear PK of filapixant [3], which allows to predict multiple dose effects based on single dose data. The fixed sequence design was chosen based on operational considerations. Considering the limited study length and that PK data are not subject to placebo effects, this was considered acceptable. The restriction of the study population to males only was considered necessary since no reproduction toxicity data were available at the time of study start. On the other hand, it is unlikely that the inclusion of women into the study would have led to relevantly different results, considering the known abundance of CYP3A4 in males and females.
4. Materials and Methods
4.1. In Vitro Studies Investigating Metabolism and Metabolizing Enzymes of Filapixant
4.1.1. Biotransformation of Filapixant in Human Liver Microsomes and Hepatocytes
[^3^H]filapixant was incubated with liver microsomes of different animal species and men as well as hepatocytes suspension of men, rat, mouse, dog, rabbit, and monkey at a concentration of 1 µM (Appendix C). The incubations were analyzed by high-performance liquid chromatography (HPLC) with tandem mass spectroscopy (MS/MS) followed by off-line radioactivity detection to generate metabolite profiles and elucidate or confirm the structures of the metabolites formed.
4.1.2. CYP Phenotyping Studies in Human Hepatocytes, Liver Microsomes, and Recombinant Human CYP Enzymes
The overall turnover of [^3^H]filapixant and formation of individual metabolites was investigated using incubations with human liver microsomes and hepatocytes (Appendix C), and recombinant human CYP isoforms (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 2J2, 3A4, 3A5, 3A7, 4A11, 4F2, 4F3A, 4F3B, 4F12, and 19A1 [aromatase]) (Table 1) at 1 µM and with CYP1A1, CYP2D6, CYP2J2, and CYP3A4 in addition at 0.1 µM. Incubations of 1 µM [^3^H]filapixant were performed in human liver microsomes in the presence or absence of CYP isoform–selective inhibitors. Drug depletion and formation of metabolites was determined in the presence of strong CYP3A4 inhibitors (itraconazole, mibefradil and azamulin) as well as other CYP isoform–selective inhibitors (1A1:alpha naphtoflavone, 7 hydroxyflavone, 2D6: quinidine, 2J2: HET0016, and telmisartan). Furthermore, 1 µM non-labeled filapixant was incubated in human hepatocytes in the presence and absence of CYP isoform–selective inhibitors, mibefradil and itraconazole. The incubations were analyzed after the fractionated collection by HPLC and posterior radioactivity detection for [^3^H]filapixant and its metabolites. When unlabeled filapixant was used, the incubation samples were analyzed by HPLC-MS/MS. Further details on the in vitro studies investigating metabolism and metabolizing enzymes of filapixant can be found in an online Supplementary Materials.
4.2. Clinical Study
The primary objective of the study was to investigate the influence of orally administered itraconazole, a strong CYP3A4/P-gp inhibitor, on the PK of orally administered filapixant. This was achieved by comparing the endpoints AUC and C_max_ of filapixant from both study periods. The other study objective was to investigate the safety and tolerability of filapixant (with/without itraconazole) in healthy male participants by investigating the frequency and intensity of TEAEs in both periods. Spontaneous reported adverse events were recorded as well adverse events that were reported after open-ended and non-leading verbal questioning of the participants to avoid bias. In addition, itraconazole concentrations were measured in plasma at selected time points to confirm that relevant inhibitory plasma levels were achieved.
The study (NCT03789890; URL: https://clinicaltrials.gov/study/NCT03789890; first registration 27 December 2018) was conducted at CRS Clinical Research Services Berlin GmbH in accordance with the ethical principles that have their origin in the Declaration of Helsinki and the International Council for harmonization (ICH) guideline E6: Good Clinical Practice (GCP) and met all local legal and regulatory requirements. The study was approved by the Ethics Committee of the State of Berlin (Ethikkommission des Landes Berlin, Fehrbelliner Platz 1,10707 Berlin, Germany) (protocol code 19431; version 2.0 from 3 December 2018; approval date 17 December 2018). Informed consent was obtained from all subjects involved in the study.
The study was conducted in a single-center, open-label, fixed sequence design with two periods. In period 1, participants received a single dose of 80 mg filapixant. After a washout period of at least 7 days to ensure the complete elimination of filapixant, period 2 started with a 14-day course of 200 mg itraconazole (Semepra^®^ liquid 10 mg/mL oral solution) once daily. On the 4th day of itraconazole treatment in period 2, participants additionally received a single dose of 80 mg filapixant, the anticipated therapeutic dose, approximately 60 min after itraconazole intake on this day. A 3-day pre-treatment period of itraconazole was chosen to reach steady state itraconazole plasma concentrations at the time of co-administration of filapixant (Figure 5). Administration of filapixant in period 1 and 2 was performed in fasted state.
Healthy male participants, aged between 18 and 55 years and with a body weight of at least 50 kg, were included. Participants’ health was assessed by the investigator (including assessment of medical history, physical examination, blood pressure, pulse rate, 12-lead electrocardiogram, body temperature, and clinical laboratory). Participants with contraindications to itraconazole and participants using any systemic or topically active medication or herbal remedies within 1 week prior to the first drug administration or during the trial until follow-up were excluded. The occasional use of ibuprofen was permissible. A full list of in- and exclusion criteria can be found in Appendix A.
Blood samples for determination of filapixant concentrations were taken from pre-dose until 96 h after administration of filapixant in Period 1 and from pre-dose until 11 days (264 h) after administration of filapixant in Period 2 (filapixant sampling: pre-dose, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12 15, 24, 36, 48, 96, 120, 144, 168, 192, 216, 240, and 264 h after drug administration; itraconazole sampling: before doses 1, 2, 3, 4, 6, 8, 12; 1, 2, and 4 h after the 4th dose and 24 h after the 14th, i.e., last, dose). Concentrations of filapixant and itraconazole were determined according to a method described before [3,8]. The method validation and analysis of the study samples were performed in compliance with the Standard Operating Procedures (SOPs) based on the EMA Guideline on Bioanalytical Method Validation, the FDA Guideline on Bioanalytical Method Validation, the Reflection Paper for Laboratories that Perform the Analysis or Evaluation of Clinical Trial Samples, and the regulations in Good Laboratory Practice (GLP) and GCP, respectively [9,10,11,12,13].
The calibration range was from 0.100 µg/L (LLOQ) to 200 µg/L (upper limit of quantification (ULOQ)) for filapixant and from 1.00 µg/L (LLOQ) to 1000 µg/L (ULOQ) for itraconazole. Accuracy (calculated as percent of nominal) and precision (CV) are as shown in Table 4.
All samples were stored at −20 °C and analyzed within 82 days after sample collection. The available stability data indicated that the analytes are stable for this time period.
Safety and tolerability were assessed based on the frequency and severity of TEAEs. Other safety assessments included standard clinical laboratory tests, vital signs, electrocardiograms, and physical examinations.
PK parameters were calculated by a non-compartmental analysis using the program WinNonlin version 5.3 (Pharsight Corporation, St. Louis, MO, USA), with the Automation Extension (version 2.90, Bayer Pharma AG, Wuppertal, Germany). The statistical evaluation was performed by using the software package SAS release 9.2 (SAS Institute Inc., Cary, NC, USA). All analyses were exploratory. Therefore, no multiplicity adjustments were performed.
The main PK metrics AUC and C_max_ were statistically analyzed, assuming log-normally distributed data. Mean differences between treatments (and their standard error) were determined for logarithmized PK metrics. A total of 90% CIs for treatment differences were determined as well, applying standard methodology for log-normally distributed data. Point estimates and exploratory 90% CIs for the ratios “filapixant + itraconazole/filapixant only” were calculated by re-transformation of the logarithmic data. Only participants providing a valid PK profile from both study periods were to be included in the primary analysis.
This was an exploratory study, and no formal statistical sample size estimation has been performed. However, a sample size of 14 participants was considered sufficient even in a scenario with high intraindividual variability of 30% to estimate the interaction effect with sufficient precision, i.e., that the 90% CIs lies within the range of 83–120% of the point estimate. This accuracy allows us to make recommendations for dosing of filapixant in future studies.
5. Conclusions
Metabolism by CYP3A4 was found to be the main elimination pathway for filapixant in vitro. Drug depletion and formation of metabolites were inhibited in the presence of strong CYP3A4 inhibitors.Filapixant was found to be a moderately sensitive substrate of CYP3A4/P-gp. Single oral dosing of 80 mg filapixant concomitantly with multiple daily oral doses of 200 mg itraconazole, a strong CYP3A4/P-gp inhibitor, led to a 1.89-fold higher C_max_ and 4.01-fold higher AUC of filapixant compared to the exposure without itraconazole co-administration. Additionally, the geometric mean terminal half-life (t_1/2_) of filapixant after a single-dose administration was prolonged by co-administration with itraconazole. The changes in both C_max_ and half-life, together with the overall change in AUC, indicate that both clearance and bioavailability were affected by co-administration of itraconazole.Single dose oral administration of 80 mg filapixant, with and without itraconazole, was well tolerated by all participants.For subsequent studies, the co-administration of filapixant at therapeutic doses with strong inhibitors of CYP3A4/P-gp should be avoided, or an appropriate dose adjustment needs to be implemented.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Morice A. Smith J.A. Mc Garvey L. Birring S.S. Parker S.M. Turner A. Hummel T. Gashaw I. Fels L. Klein S. Eliapixant (BAY 1817080), a P 2X 3 receptor antagonist, in refractory chronic cough: A randomised, placebo-controlled, crossover phase 2a study Eur. Respir. J.202158200424010.1183/13993003.04240-202033986030 PMC 8607926 · doi ↗ · pubmed ↗
- 2Friedrich C. Francke K. Birring S.S. Van Den Berg J.W.K. Marsden P.A. Mc Garvey L. Turner A.M. Wielders P. Gashaw I. Klein S. The P 2X 3 Receptor Antagonist Filapixant in Patients with Refractory Chronic Cough: A Randomized Controlled Trial Respir. Res.20232410910.1186/s 12931-023-02384-837041539 PMC 10088222 · doi ↗ · pubmed ↗
- 3Friedrich C. Singh D. Francke K. Klein S. Hetzel T. Zolk O. Gashaw I. Scheerans C. Morice A. Pharmacodynamics, Pharmacokinetics and CYP 3A 4 Interaction Potential of the Selective P 2X 3 Receptor Antagonist Filapixant: A Randomized Multiple Ascending-dose Study in Healthy Young Men Br. J. Clin. Pharmacol.2024902004201810.1111/bcp.1609138775025 · doi ↗ · pubmed ↗
- 4European Medicines Agency Guideline on the Investigation of Drug Interactions 2012 Available online: www.ema.europa.eu/contact(accessed on 1 June 2025)
- 5Food and Drug Administration Clinical Drug Interaction Studies-Cytochrome P 450 Enzyme-and Transporter-Mediated Drug Interactions Guidance for Industry 2020 Available online: https://www.fda.gov/Drugs/Guidance Compliance Regulatory Information/Guidances/default.htm(accessed on 1 June 2025)
- 6Hardin T.C. Graybill J.R. Fetchick R. Woestenborghs R. Rinaldi M.G. Kuhn J.G. Pharmacokinetics of Itraconazole Following Oral Administration to Normal Volunteers Antimicrob. Agents Chemother.1988321310131310.1128/AAC.32.9.13102848442 PMC 175857 · doi ↗ · pubmed ↗
- 7Klein S. Gashaw I. Baumann S. Chang X. Hummel T. ThußU. Friedrich C. First-in-human Study of Eliapixant (BAY 1817080), a Highly Selective P 2X 3 Receptor Antagonist: Tolerability, Safety and Pharmacokinetics Br. J. Clin. Pharmacol.2022884552456410.1111/bcp.1535835437837 PMC 9546310 · doi ↗ · pubmed ↗
- 8Friedrich C. Francke K. Gashaw I. Scheerans C. Klein S. Fels L. Smith J.A. Hummel T. Morice A. Safety, Pharmacodynamics, and Pharmacokinetics of P 2X 3 Receptor Antagonist Eliapixant (BAY 1817080) in Healthy Subjects: Double-Blind Randomized Study Clin. Pharmacokinet.2022611143115610.1007/s 40262-022-01126-135624408 PMC 9349145 · doi ↗ · pubmed ↗