Asiatic Acid from Centella asiatica as a Potent EGFR Tyrosine Kinase Inhibitor with Anticancer Activity in NSCLC Cells Harboring Wild-Type and T790M-Mutated EGFR
Chaiwat Monmai, Sahachai Sabuakham, Wachirachai Pabuprapap, Waraluck Chaichompoo, Apichart Suksamrarn, Panupong Mahalapbutr

TL;DR
Asiatic acid from Centella asiatica shows strong anticancer activity by inhibiting EGFR in lung cancer cells, including those with a resistant mutation.
Contribution
Asiatic acid is identified as a novel EGFR tyrosine kinase inhibitor effective against both wild-type and T790M-mutated EGFR.
Findings
Asiatic acid potently inhibits wild-type and double-mutant EGFR with better binding energy than existing drugs.
Asiatic acid reduces cancer cell viability and induces apoptosis without harming normal lung cells.
Asiatic acid suppresses ERK and Akt signaling pathways and inhibits EGFR activation in lung cancer cells.
Abstract
Lung cancer is a leading cause of cancer mortality worldwide. Targeted therapies with epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) represent a significant advance in the management of lung cancer. However, their long-term efficacy is often limited by acquired resistance, particularly due to the T790M mutation, highlighting the need for novel EGFR-TKIs. Although compounds derived from Centella asiatica have demonstrated anticancer potential, their role in EGFR inhibition has not yet been reported. In this study, we investigated the inhibitory activity of two primary constituents, asiaticoside and asiatic acid, against wild-type and double-mutant (L858R/T790M) EGFR, as well as the anticancer effects of the more potent compound in lung cancer cells. A kinase activity assay revealed that asiatic acid potently inhibited both wild-type and double-mutant EGFR,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
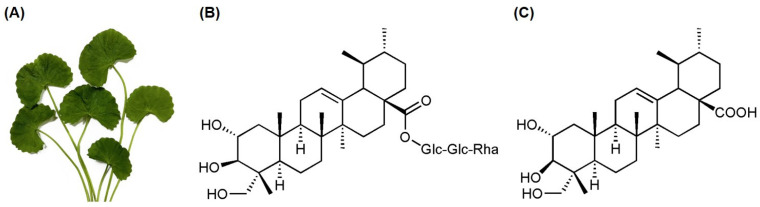 Figure 1
Figure 1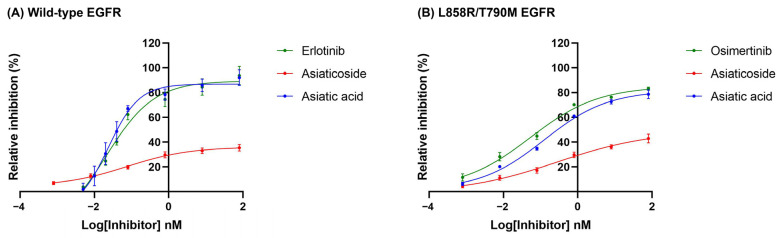 Figure 2
Figure 2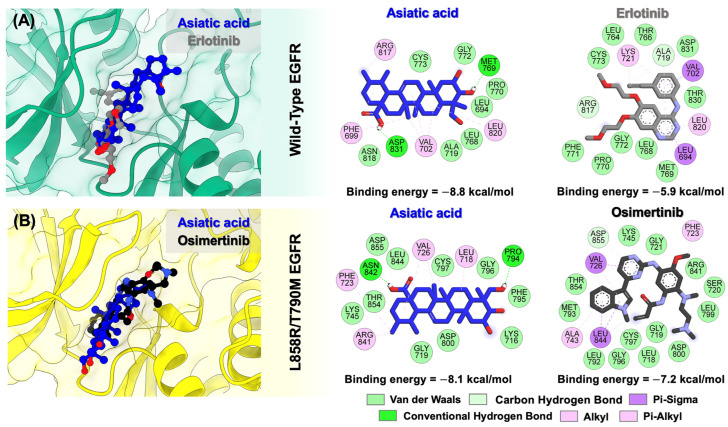 Figure 3
Figure 3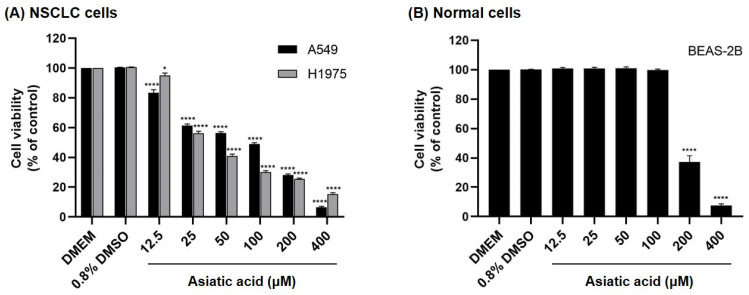 Figure 4
Figure 4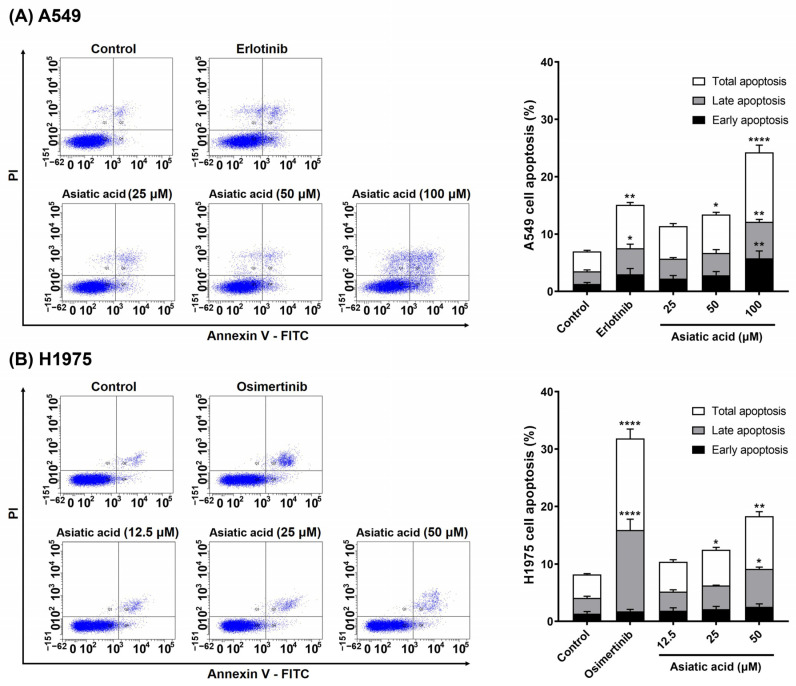 Figure 5
Figure 5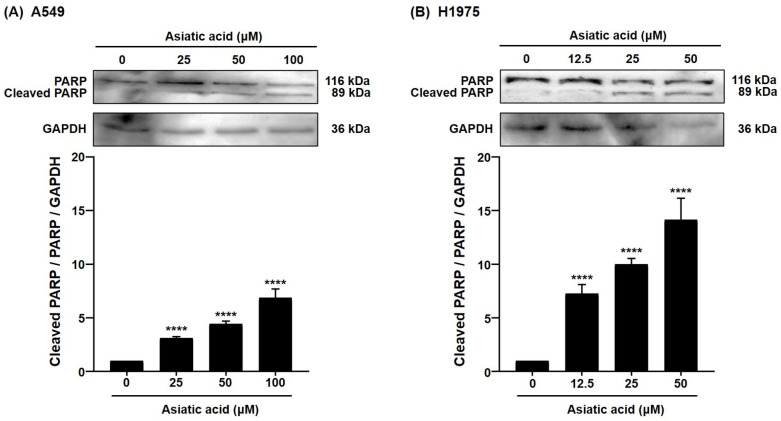 Figure 6
Figure 6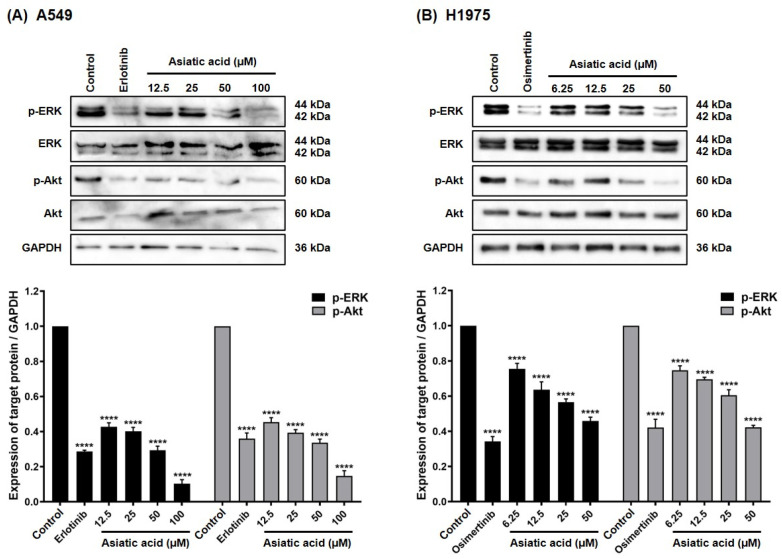 Figure 7
Figure 7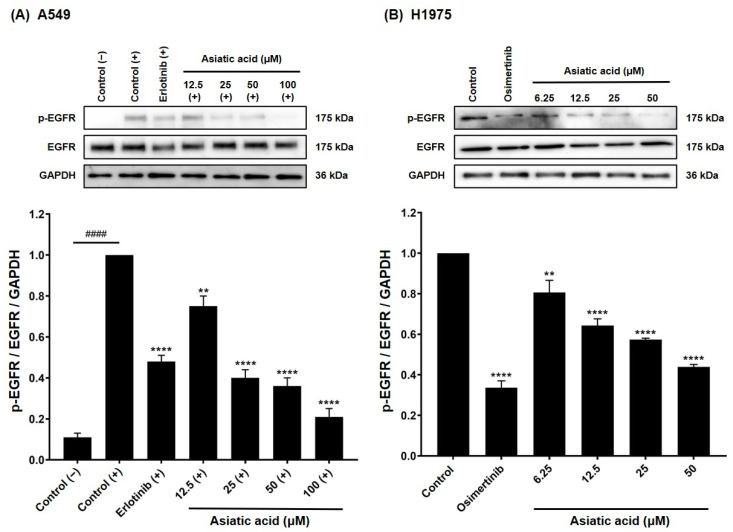 Figure 8
Figure 8- —Fundamental Fund of Khon Kaen University
- —Post Doctoral Training Program for Frontier Research from Khon Kaen University, Thailand
- —Center of Excellence for Innovation in Chemistry, Ministry of Higher Education, Science, Research and Innovation, Thailand
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMedicinal Plants and Neuroprotection · Advanced Nanomaterials in Catalysis · Scientific and Engineering Research Topics
1. Introduction
Cancer, a multifaceted and devastating group of diseases, poses a major threat to global public health. It is characterized by the unchecked growth and spread of abnormal cells, which disrupt normal tissue function and can be fatal if left untreated [1,2]. The scale of the problem is staggering: nearly 20 million new cases and about 10 million deaths are reported each year, and these figures are projected to rise, underscoring the urgent need for more effective prevention, diagnosis, and treatment strategies [3]. Among these, lung cancer remains a major global health challenge and one of the leading causes of cancer-related mortality worldwide [4]. In 2020, it accounted for approximately 2.21 million new cases and 1.80 million deaths, making it the second most common cancer [5,6].
Epidermal growth factor receptor (EGFR) plays a crucial role in regulating cell proliferation, differentiation, and survival [7]. Autophosphorylation of EGFR triggers multiple downstream signaling pathways, including mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK), phosphatidylinositol 3-kinase/Akt (PI3K/Akt), and Janus kinase/signal transducer and activator of transcription (JAK/STAT) [8]. Alterations in EGFR—such as overexpression, exon 19 deletions, and the L858R substitution in exon 21—are key oncogenic drivers in non-small cell lung cancer (NSCLC) [9]. Erlotinib, a first-generation EGFR tyrosine kinase inhibitor (EGFR-TKI), provides clinical benefits to NSCLC patients [10]. However, its long-term efficacy is often limited by acquired resistance, particularly due to the T790M mutation [11]. Consequently, the development of novel EGFR-TKIs remains a critical need.
Centella asiatica (Figure 1A) is widely used as a medicinal herb across Asia, Europe, Australia, the Americas, and Southern Africa [12]. It exhibits a wide range of pharmaceutical potentials, including treatment of various skin conditions [13], promotion of wound healing [14], antioxidant [15], anti-inflammatory [16], and anti-apoptotic [17] activities. The primary active constituents of C. asiatica are pentacyclic triterpenes [18], including asiaticoside [19], asiatic acid [20,21,22], madecassoside [23], and madecassic acid [24], which have demonstrated anticancer potential. Although the anticancer effects of C. asiatica’s active compounds have been studied, their targeted inhibition of EGFR (both wild-type and T790M mutant) has not yet been reported. In this study, we evaluated the inhibitory activity of two major constituents, asiaticoside and asiatic acid, against both wild-type and double-mutant (L858R/T790M) EGFR, and further examined the anticancer effects of the more potent compound in human NSCLC cell lines expressing wild-type EGFR (A549) and double-mutant EGFR (H1975).
2. Materials and Methods
2.1. Extraction and Isolation of Asiaticoside and Asiatic Acid
Centella asiatica (Apiaceae) was collected from Nonthaburi Province, Thailand, in 2022. The plant species was identified by Mr. Yanyong Punpreuk, Department of Agriculture, Bangkok, Thailand, and a voucher specimen was deposited at the Faculty of Science, Ramkhamhaeng University (Apichart Suksamrarn, No. 102). Asiaticoside and asiatic acid were isolated from the aerial parts of this plant species. Briefly, the fresh aerial parts of C. asiatica (1.3 kg) were washed, air-dried, milled, and macerated with MeOH. The filtered solution was evaporated under reduced pressure at 40–45 °C to yield the MeOH extract (75.8 g). The crude extract was subjected to column chromatography on silica gel using a CH_2_Cl_2_–MeOH gradient system, yielding five fractions. Fraction 2 was chromatographed on silica gel column (CH_2_Cl_2_–MeOH, 95:5) to yield asiatic acid (28.5 mg, 0.04% yield). Fraction 4 was purified by reverse phase-C_18_ column chromatography (MeOH–H_2_O, 1:4) to afford asiaticoside (142.8 mg, 0.19% yield). The structures of asiaticoside and asiatic acid were confirmed by comparing their spectroscopic data (^1^H, ^13^C NMR, and mass spectra; Figures S1–S6) with those previously reported in the literature ([25] for asiaticoside and [26] for asiatic acid). The purity level of the isolated compounds is greater than 95% from ^1^H NMR and TLC investigations. Compounds with high molecular weight typically exhibit poor solubility, which can be attributed to their stronger intermolecular interactions, greater chain entanglement, and larger non-polar surface area [27]. Therefore, the stock solutions of asiaticoside and asiatic acid were prepared at a concentration of 50 mM in 100% DMSO.
2.2. EGFR Inhibitory Activity of Asiaticoside and Asiatic Acid
The EGFR inhibitory activity of the tested compound was measured using the ADP-Glo^TM^ assay (Promega, WI, USA) according to a previous study [28]. Kinase reactions were performed in a 384-well plate and contained 2 μL of the tested compound, 8 μL of 1X kinase buffer, 25 μM ATP, 2.5 μM Poly(Glu-Tyr), and EGFR enzyme [1.25 ng/μL; wild-type (SRP0239), or L858R/T790M (SRP0242); Sigma-Aldrich, St. Louis, MO, USA]. For the positive control, 2 μL of DMSO was added in place of the tested compound. For the negative control, the EGFR enzyme was omitted and replaced with 5 μL of 1X kinase buffer. The plate was protected from light and incubated at room temperature for 1 h, followed by the addition of 5 μL ADP-Glo^TM^ reagent and a further 40-min incubation. To convert ADP to ATP, 10 μL of kinase detection reagent was added, followed by a 30-min incubation under the same conditions. ATP levels were then measured using a Varioskan LUX multimode microplate reader (Thermo Fisher Scientific, Waltham, MA, USA), and percent inhibition was calculated using Equation (1).
The IC_50_ of each compound was calculated using GraphPad Prism software (version 8.0.2, GraphPad Software, Inc., Boston, MA, USA).
2.3. Preparation of Structure and Molecular Docking
The binding interactions of the studied compound with wild-type and double-mutant EGFR were evaluated using molecular docking. The crystal structures of EGFR were retrieved from the Protein Data Bank (PDB), using PDB entry 1M17 for wild-type EGFR and 4I22 for the double-mutant (L858R/T790M) EGFR. The three-dimensional (3D) structure of the tested compound was constructed using the GaussView 6.0 program (Gaussian, Inc., Wallingford, CT, USA). Protonation states of the protein and ligand at physiological pH (7.4) were assigned using the APBS webserver (https://server.poissonboltzmann.org, accessed on 7 August 2025) and MarvinSketch (version: 24.3.0: Chemaxon Ltd., Budapest, Hungary), respectively. The optimized protein and ligand structures were converted to pdbqt format using AutoDockTools (version 1.5.7; Molecular Graphics Laboratory, The Scripps Research Institute, La Jolla, CA, USA). The grid box dimensions and centers were defined as follows: for wild-type EGFR, the grid size was set to 50 Å × 50 Å × 60 Å with center coordinates at x = 25.32, y = −0.99, and z = 53.94; for double-mutant EGFR, the grid size was 50 Å × 50 Å × 50 Å with center coordinates at x = 10.66, y = −15.55, and z = 8.89. Molecular docking was performed using AutoDock version 4.2.6, and the docking energies (kcal/mol) of the resulting protein-ligand complexes were obtained using a Lamarckian genetic algorithm and an empirical binding free energy function. To validate the docking protocol, the co-crystallized ligand was re-docked, yielding RMSD values of 1.69 Å for wild-type EGFR and 1.25 Å for mutant EGFR (Figure S7), confirming the accuracy of the procedure. Two-dimensional (2D) interaction profiles of the studied compounds with their target proteins were generated using Discovery Studio Visualizer (version 21.1.0.20298; BIOVIA, San Diego, CA, USA).
2.4. Cell Lines and Cell Culture
BEAS-2B, A549, and H1975 cell lines were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). All cancer cell lines (A549 and H1975) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) antibiotic-antimycotic (penicillin, streptomycin, and amphotericin B; Gibco, Brooklyn, NY, USA), at 37 °C in a 5% CO_2_ humidified incubator. BEAS-2B cells (a non-cancerous human bronchial epithelium) were maintained in Airway Epithelial Cell Medium (AECM; PCS-300-030, ATCC) supplemented with the Bronchial Epithelial Cell Growth Kit (PCS-300-040, ATCC) under identical incubation conditions.
2.5. Cell Viability Assay
Cells were seeded in a 96-well plate at 1,500 cells/well and incubated at 37 °C for 24 h. After incubation, the culture medium was replaced with fresh medium containing various concentrations of asiatic acid. Cells treated with 0.8% (v/v) DMSO served as the vehicle control. After 72 h, cells were incubated with 100 μL of MTT solution (0.5 mg/mL in PBS) for 4 h at 37 °C. The solution was then removed, 100 μL of DMSO was added, and the plate was protected from light with gentle agitation for 15 min. Absorbance was recorded at 540 nm using a Sunrise microplate reader (Tecan Group Ltd., Männedorf, Switzerland).
2.6. Cell Apoptosis Assay
Cells were plated in a 6-well plate at a density of 200,000 cells/well and allowed to adhere overnight at 37 °C. The following day, cells were treated with increasing concentrations of asiatic acid for 24 h. Erlotinib (10 μM) [29,30] and osimertinib at 5 μM (Table S1) were used as reference drugs for A549 and H1975 cells, respectively. After treatment, cells were harvested by trypsinization and centrifugation. The cell pellets were stained with FITC-conjugated Annexin V (640945; BioLegend, San Diego, CA, USA) and incubated for 30 min at room temperature in the dark. Propidium iodide (PI; P3566; Invitrogen^TM^, Waltham, MA, USA) was then added, and apoptotic populations were analyzed using a BD FACSCalibur flow cytometer (BD Biosciences, Heidelberg, Germany).
2.7. Western Blot Analysis
Cells were plated at 200,000 cells/well in a 6-well plate and allowed to adhere for 24 h. They were then treated with the tested compound at varying concentrations for 24 h. For A549 cells, treatment was performed in serum-free medium for 24 h, followed by stimulation with 100 ng/mL human EGF for 10 min prior to harvesting [31,32]. Cells were harvested by trypsinization, lysed in RIPA buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 1% Na-deoxycholate, and 0.1% SDS] with protease and phosphatase inhibitors, and incubated on ice for 30 min. Lysates were centrifuged, and protein concentration was determined using the Bradford assay. Equal amounts (30 μg) were separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane (Immobilon^®^-P, Merck Millipore, Burlington, MA, USA). The membrane was blocked with 5% skim milk in 1X TBST and incubated overnight at 4 °C with primary antibodies (Table S2) diluted 1:1000 in 1% skim milk-TBST or 1% BSA-TBST. After washing, membranes were incubated at room temperature for 1 h with secondary antibodies—goat anti-Rabbit IgG or goat anti-Mouse IgG (Invitrogen, Waltham, MA, USA)—diluted 1:2000. Protein signals were detected using Luminata^TM^ Crescendo Western HRP Substrate (Merck Millipore) and imaged with an ImageQuant 800 system (GE Healthcare Bio-Sciences, Uppsala, Sweden). The auto exposure mode was selected to automatically adjust the exposure time to prevent image saturation. The detected signal was quantified using Image Studio Lite (version 5.2, LICORbio^TM^, Lincoln, NE, USA).
2.8. Statistic Analysis
Data are presented as mean ± standard error of the mean (SEM) from three independent experiments. Statistical differences between groups were assessed using one-way ANOVA followed by Tukey–Kramer post hoc test. Differences with p < 0.05 were considered statistically significant.
3. Results
3.1. Inhibitory Effect of Asiaticoside and Asiatic Acid on the EGFR Tyrosine Kinase Activity
To determine the EGFR inhibitory activity of the two major compounds isolated from C. asiatica, the ADP-Glo^TM^ assay was performed. Erlotinib is a reversible EGFR-TKI that blocks receptor activity by occupying the ATP-binding pocket of the kinase domain [33]. In contrast, osimertinib is a third-generation, irreversible EGFR-TKI that selectively targets the T790M resistance mutation by covalently binding to cysteine 797 within the ATP-binding pocket of the mutant receptor [34]. Accordingly, erlotinib and osimertinib were used as positive controls for wild-type and T790M-mutant EGFR, respectively.
As shown in Figure 2, asiaticoside, asiatic acid, and the known EGFR-TKIs inhibited the kinase activity of both wild-type and double-mutant (T790M/L858R) forms of EGFR in a concentration-dependent manner. The IC_50_ values for kinase inhibition against wild-type EGFR were as follows: erlotinib, 0.062 ± 0.009 nM; asiaticoside, >80 nM; and asiatic acid, 0.035 ± 0.002 nM. Asiatic acid exhibited the lowest IC_50_ value, indicating the highest EGFR kinase inhibitory activity. For the double-mutant EGFR, the IC_50_ values were: osimertinib, 0.116 ± 0.038 nM; asiaticoside, >80 nM; and asiatic acid, 0.348 ± 0.055 nM (Table 1). Based on these results, only asiatic acid was chosen for further experiments due to its stronger inhibitory activity against both wild-type and mutant EGFR compared to asiaticoside.
The inhibitory effect of asiatic acid on fibroblast growth factor receptor (FGFR) was further examined to evaluate its selectivity. Results showed that asiatic acid inhibited FGFR kinase activity, with an IC_50_ of 0.471 nM (Figure S8). Overall, asiatic acid exhibits highly potent EGFR kinase inhibition and demonstrates approximately 13.5-fold greater selectivity for EGFR over FGFR.
3.2. Molecular Docking of Asiatic Acid into the Wild-Type and Double-Mutant Forms of EGFR
To elucidate how asiatic acid interacts with wild-type and mutant EGFR, molecular docking was performed. As shown in Figure 3, superimposition revealed that asiatic acid bound to the ATP-binding pocket of both wild-type EGFR and L858R/T790M EGFR similar to the known drugs, erlotinib and osimertinib. For wild-type EGFR, asiatic acid and erlotinib formed complexes with binding energies of −8.8 and −5.9 kcal/mol, respectively, whereas in the L858R/T790M mutant EGFR, asiatic acid and osimertinib bound with energies of −8.1 and −7.2 kcal/mol, respectively.
The complexation between asiatic acid and the wild-type EGFR was primarily stabilized by van der Waals and alkyl/pi-alkyl interactions with LEU694, PHE699, VAL702, ALA719, LEU768, PRO770, GLY772, CYS773, ARG817, ASN818, and LEU820 residues. Moreover, asiatic acid formed two hydrogen bonds with MET769 and ASP831 residues. Similarly, erlotinib binding to wild-type EGFR was mainly stabilized through van der Waals and pi interactions. Notably, several residues—LEU694, VAL702, ALA719, LEU768, MET769, PRO770, GLY772, CYS773, ARG817, LEU820, and ASP831—were common to both the asiatic acid and erlotinib complexes.
For double-mutant EGFR, asiatic acid formed several van der Waals interactions with LYS716, GLY719, LYS745, PHE795, GLY796, CYS797, ASP800, LEU844, THR854, and ASP855; alkyl/pi-alkyl interactions with LEU718, PHE723, VAL726, and ARG841; and hydrogen bonds with PRO794 and ASN842. Similarly, osimertinib binding to double-mutant EGFR was mainly stabilized through van der Waals and pi interactions. However, hydrogen bond formation was not observed in the osimertinib complex. The amino acid residues commonly involved in both asiatic acid–L858R/T790M EGFR and osimertinib–L858R/T790M EGFR complexes include LEU718, GLY719, PHE723, VAL726, LYS745, Gly796, CYS797, ASP800, ARG841, LEU844, THR854, and ASP855.
3.3. Effect of Asiatic Acid on the Cell Viability
The cytotoxicity of asiatic acid was assessed in A549 (expressing wild-type EGFR) and H1975 (expressing L858R/T790M EGFR) [35] NSCLC cells using the MTT assay. As shown in Figure 4, asiatic acid suppressed cell viability in a concentration-dependent manner and exhibited strong cytotoxicity against A549 and H1975 cells, with IC_50_ values of 64.52 ± 2.49 μM and 36.55 ± 0.86 μM, respectively (Table 2). In contrast, asiatic acid displayed low cytotoxicity in non-cancerous BEAS-2B lung epithelial cells, with significant effects only at concentrations >100 μM (IC_50_ = 184.10 ± 5.38 μM), while concentrations ≤100 μM showed no notable cytotoxicity.
Compared with A549 cells, H1975 cells showed a more pronounced inhibitory response, in which asiatic acid treatment at concentrations of ≥100 μM reduced cell viability to below 40%. Based on these findings, concentrations of up to 100 μM for A549 cells and 50 μM for H1975 cells were selected for subsequent experiments.
3.4. Effect of Asiatic Acid on the A549 and H1975 Cell Apoptosis
To evaluate the role of apoptosis in asiatic acid-mediated cytotoxicity in NSCLC cells, Annexin V/PI staining was analyzed by flow cytometry. As shown in Figure 5, asiatic acid treatment significantly increased apoptosis in both A549 and H1975 cells compared to the vehicle control. At the highest tested concentrations, the apoptotic cell population increased approximately threefold in A549 cells (100 μM) and about twofold in H1975 cells (50 μM) relative to the DMSO-treated group. In A549 cells, 100 μM asiatic acid significantly induced both early (p = 0.0093) and late apoptosis (p = 0.0040), while in H1975 cells, 50 μM asiatic acid predominantly triggered late-stage (p = 0.0316) apoptosis. For known EGFR-TKIs, erlotinib treatment (10 μM) increased apoptosis approximately twofold in A549 cells, while osimertinib (5 μM) led to a threefold increase in apoptosis in H1975 cells compared to the untreated group.
The expression of cleaved poly(ADP-ribose) polymerase (PARP), a hallmark of programmed cell death, was further examined in A549 and H1975 cells to confirm the apoptosis-inducing activity of asiatic acid in NSCLC cells. As shown in Figure 6, treatment with asiatic acid led to a significant, concentration-dependent increase in cleaved PARP (Asp214) levels in A549 cells (25–100 μM, p < 0.0001). Similarly, in H1975 cells, cleaved PARP expression was significantly elevated following asiatic acid treatment (12.5–50 μM, p < 0.0001). These findings indicate that asiatic acid promotes PARP cleavage in both cell lines, consistent with its apoptosis-inducing activity (Figure 5).
3.5. Effect of Asiatic Acid on EGFR Downstream Signaling Pathways
Western blot analysis was conducted to examine the effect of asiatic acid on EGFR-mediated survival pathways. As shown in Figure 7, phosphorylated ERK and Akt [p-ERK (Thr202/Tyr204) and p-Akt (Ser473)] were detected in both A549 and H1975 cells. Treatment with erlotinib (10 μM) and osimertinib (5 μM) significantly (p < 0.0001) reduced p-ERK and p-Akt levels in A549 and H1975 cells, respectively. In A549 cells, asiatic acid significantly decreased p-ERK and p-Akt levels compared to the vehicle control, with 100 μM treatment reducing these proteins more than erlotinib. Similarly, in H1975 cells, asiatic acid caused a significant, dose-dependent decrease in p-ERK and p-Akt (p < 0.0001). These results demonstrate that asiatic acid inhibits ERK and Akt signaling pathways in both NSCLC cell lines.
3.6. Effect of Asiatic Acid on EGFR Activation in A549 and H1975 Cells
To assess whether asiatic acid–induced reductions in p-ERK and p-Akt were mediated through EGFR inhibition, the levels of p-EGFR (Tyr1068) were examined using Western blot. In A549 cells, basal p-EGFR expression was initially low (Figure 8A). Stimulation with human EGF increased p-EGFR levels, confirming activation of the EGFR. Treatment with 10 μM erlotinib significantly reduced EGF-induced EGFR phosphorylation compared with the EGF-stimulated group (p < 0.0001). Likewise, asiatic acid significantly inhibited EGF-induced EGFR phosphorylation in a concentration-dependent fashion. In H1975 cells, which harbor constitutively active double-mutant EGFR, basal p-EGFR expression was high without stimulating with EGF (Figure 8B). Treatment with 5 μM osimertinib significantly suppressed (p < 0.0001) p-EGFR levels compared to the control. Similarly, asiatic acid significantly reduced p-EGFR levels in H1975 cells in a dose-dependent manner.
Furthermore, the cellular thermal shift assay (CETSA) revealed that treatment with 100 μM asiatic acid markedly enhanced the stability of the EGFR protein in A549 cells, indicating strong binding affinity toward EGFR (Figure S9). Collectively, these findings indicated that asiatic acid effectively suppresses EGFR activation in both A549 and H1975 NSCLC cells, highlighting its potential as a novel TKI targeting both wild-type and mutant forms of EGFR.
4. Discussion
Currently, chemotherapy is widely used as a treatment for patients with wild-type EGFR, while TKIs are employed for those with EGFR mutations [36]. However, a major issue of chemotherapy is its lack of selectivity for cancer cells, leading to significant toxicity in normal cells [37]. Furthermore, acquired resistance to TKIs caused by the T790M mutation in the EGFR poses a significant therapeutic challenge [11]. Together, these issues—non-selective cytotoxicity and drug resistance—highlight the urgent need for novel anticancer agents capable of effectively targeting both wild-type and T790M-mutat EGFR.
Previous studies have shown that asiatic acid effectively inhibited the proliferation of various cancer cells, including human colon cancer cells (SW480 and HCT116) [20], human liver cancer cells (HepG2) [22], and human breast cancer cells (MCF-7 and MDA-MB-231) [21]. Additionally, asiatic acid and its derivatives have been reported to exhibit antiproliferative effect on NSCLC cells [38,39]. Consistent with these findings, our results showed that asiatic acid isolated from C. asiatica effectively inhibited the proliferation of A549 and H1975 NSCLC cells within a concentration range of 12.5–400 μM, while showing no cytotoxicity toward BEAS-2B normal lung cells at concentrations of 12.5–100 μM (Figure 4 and Table 2). Notably, H1975 cells, which harbor EGFR double mutations, were more sensitive to asiatic acid (IC_50_ = 36.55 ± 0.86 µM) compared with A549 cells expressing wild-type EGFR (IC_50_ = 64.52 ± 2.49 µM).
As shown in Figure 5, asiatic acid significantly induced apoptosis in both A549 and H1975 cells. This finding is supported by previous work from Wu et al., demonstrating that asiatic acid significantly induced apoptotic cell death in lung cancer cells both in vitro and in vivo [40]. PARP is a DNA repair protein that detects DNA damage and facilitates repair processes. During apoptosis, caspases cleave PARP into smaller fragments, and the presence of cleaved PARP serves as a molecular marker of apoptosis, reflecting the loss of PARP’s DNA repair function and the progression of programmed cell death [41,42]. Consistent with this, our results showed that asiatic acid significantly increased cleaved PARP levels in both A549 and H1975 cells (Figure 6).
Cancer progression and growth are closely associated with the activation of intracellular signaling pathways mediated by EGFR, which is overexpressed in up to 60% of NSCLC cases [43,44]. As shown in Figure 8, treatment with asiatic acid significantly suppressed EGFR activation (p-EGFR) in A549 and H1975 NSCLC cell lines. Supportively, molecular docking demonstrated that asiatic acid could bind to the ATP-binding pocket of wild-type and double-mutant EGFR with binding energies superior to those of erlotinib and osimertinib, and interacted with key active-site residues (Figure 3), leading to suppression of EGFR tyrosine kinase activity (Figure 2 and Table 1).
EGFR phosphorylation activates multiple downstream signaling pathways, including MAPK/ERK and PI3K/Akt cascades [45,46]. ERK activation, a common mechanism in many cell types, is often associated with cancer progression [47]. Overactivation of ERK and Akt, frequently triggered by p-EGFR, contributes to aggressive tumor phenotypes and therapy resistance [48]. As shown in Figure 7, A549 and H1975 cells exhibited high levels of p-ERK and p-Akt, which were significantly suppressed by treatment with asiatic acid or the known TKIs erlotinib and osimertinib. These findings align with previous reports demonstrating that asiatic acid and its analog significantly downregulated p-ERK [38] and p-Akt [49] in A549 cells.
5. Conclusions
Our findings highlight the promising anticancer potential of asiatic acid from C. asiatica, which demonstrated potent EGFR inhibitory activity, selective cytotoxicity toward NSCLC cells, apoptosis induction, and effective suppression of EGFR-mediated signaling pathways. By targeting both wild-type and mutant forms of EGFR, asiatic acid emerges as a candidate for further development as a targeted therapeutic agent for lung cancer.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Din S.R.U. Saeed S. Khan S.U. Arbi F.M. Xuefang G. Zhong M. Bacteria-driven cancer therapy: Exploring advancements and challenges Crit. Rev. Oncol. Hematol.202319110414110.1016/j.critrevonc.2023.10414137742883 · doi ↗ · pubmed ↗
- 2Anand U. Dey A. Chandel A.K.S. Sanyal R. Mishra A. Pandey D.K. De Falco V. Upadhyay A. Kandimalla R. Chaudhary A. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics Genes. Dis.2023101367140110.1016/j.gendis.2022.02.00737397557 PMC 10310991 · doi ↗ · pubmed ↗
- 3Wang Z. Liu S. Zhang M. Liu M. Dual roles of methylglyoxal in cancer Front. Oncol.202515155716210.3389/fonc.2025.155716240352588 PMC 12061732 · doi ↗ · pubmed ↗
- 4Yu X. Wang Z. Chen Y. Yin G. Liu J. Chen W. Zhu L. Xu W. Li X. The predictive role of immune related subgroup classification in immune checkpoint blockade therapy for lung adenocarcinoma Front. Genet.20211277183010.3389/fgene.2021.77183034721552 PMC 8554034 · doi ↗ · pubmed ↗
- 5Li L. Chen C. Xiang Q. Fan S. Xiao T. Chen Y. Zheng D. Transient receptor potential cation channel subfamily V member 1 expression promotes chemoresistance in non-small-cell lung cancer Front. Oncol.20221277365410.3389/fonc.2022.77365435402237 PMC 8990814 · doi ↗ · pubmed ↗
- 6Bai Y. Yang W. Käsmann L. Sorich M.J. Tao H. Hu Y. Immunotherapy for advanced non-small cell lung cancer with negative programmed death-ligand 1 expression: A literature review Transl. Lung Cancer Res.20241339842210.21037/tlcr-23-14438496691 PMC 10938091 · doi ↗ · pubmed ↗
- 7Gutierrez E. Cahatol I. Bailey C.A.R. Lafargue A. Zhang N. Song Y. Tian H. Zhang Y. Chan R. Gu K. Regulation of Rho B gene expression during tumorigenesis and aging process and its potential applications in these processes Cancers 20191181810.3390/cancers 1106081831200451 PMC 6627600 · doi ↗ · pubmed ↗
- 8Haraldsdottir S. Bekaii-Saab T. Integrating anti-EGFR therapies in metastatic colorectal cancer J. Gastrointest. Oncol.2013428529810.3978/j.issn.2078-6891.2013.02823997940 PMC 3712294 · doi ↗ · pubmed ↗