Increased EGFR/HER2 Pathway Activation Contributes to Skin Tumorigenesis in Tpl2−/− Mice
Laura R. Purkey, Stefania Mehedincu, Charles Irvine, Raelyn Akdag, Megan Little, W. Wade Kothmann, Katharine Rus, Erin Greenberg, Neil Shady, Kathleen DeCicco-Skinner

TL;DR
Removing a specific protein in mice leads to increased skin tumor growth, but blocking related proteins can significantly reduce tumors.
Contribution
This study identifies compensatory ErbB signaling as a novel driver of skin tumorigenesis in mice lacking Tpl2, suggesting new therapeutic strategies.
Findings
Tpl2−/− mice develop 12-fold more papillomas and 4-fold more cSCCs compared to wild-type mice.
Inhibiting EGFR or HER2 with Gefitinib or Lapatinib reduces tumor burden in Tpl2−/− mice.
Tpl2 loss increases EGFR, HER2, and HER3 gene expression and activates ErbB signaling in papillomas.
Abstract
The mitogen-activated protein kinase (MAPK) signaling pathway plays a critical role in cellular growth and survival, and its disruption is associated with various cancers. Loss or alteration of critical regulatory proteins in this pathway can trigger compensatory mechanisms that activate alternative signaling routes that promote tumor development. This study demonstrates that loss of a specific MAPK family member, Tpl2, leads to upregulation of growth factor receptors and associated signaling molecules, resulting in dramatically increased tumor formation in mice. Importantly, targeted inhibition of these upregulated receptors significantly reduced tumor burden, suggesting that therapeutic strategies focused on blocking compensatory signaling pathways could be valuable for treating cancers that arise from MAPK pathway dysfunction. Background: The mitogen-activated protein kinase (MAPK)…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
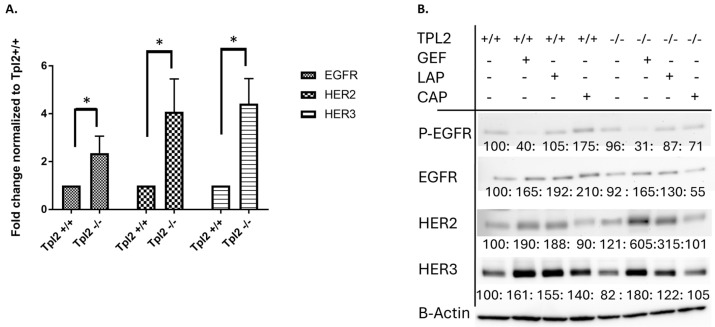 Figure 1
Figure 1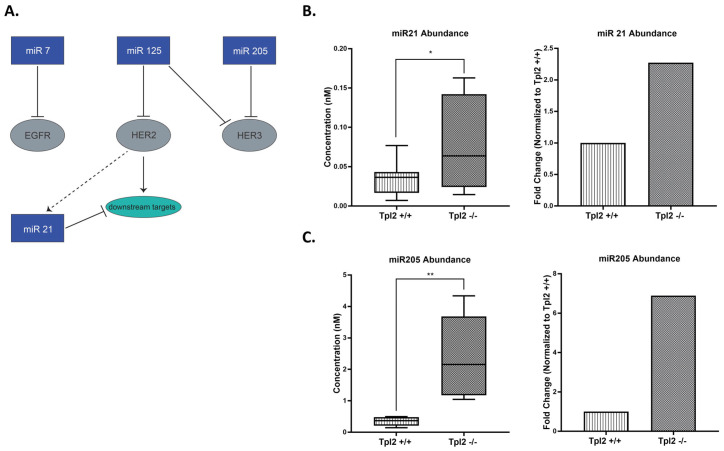 Figure 2
Figure 2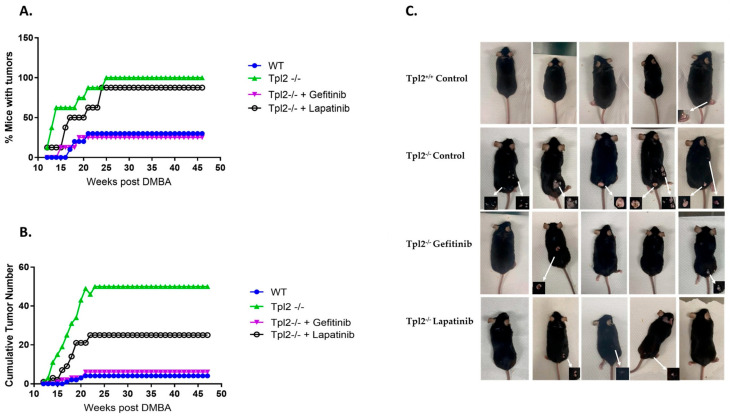 Figure 3
Figure 3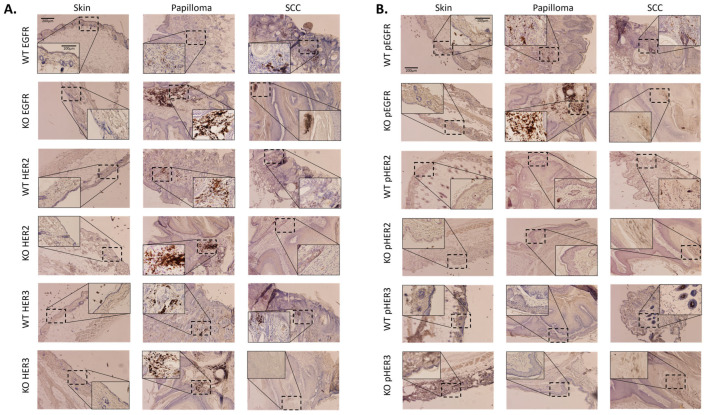 Figure 4
Figure 4- —Mellon funding (American University)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHER2/EGFR in Cancer Research · Melanoma and MAPK Pathways · Monoclonal and Polyclonal Antibodies Research
1. Introduction
Cutaneous squamous cell carcinoma (cSCC) is the second most prevalent form of skin cancer globally [1,2]. There has been a 300% increase in cSCC diagnoses over the past three decades, and this upward trajectory is projected to continue, driven by factors such as heightened exposure to ultraviolet and chemical carcinogens, as well as an aging population [2,3]. While surgery is effective for most patients with cSCC, those with metastatic and advanced cases have few treatment options with poor outcomes [4]. Further, a high mutation rate complicates the identification of key driver mutations [5].
The mitogen-activated protein kinase (MAPK) pathways, which are frequently dysregulated in cSCC, serve as essential regulators of numerous cellular processes, including proliferation, survival, differentiation, and inflammatory responses [6]. These sophisticated, interconnected networks respond to various stimuli, such as growth factors, stress signals, and inflammatory mediators [7]. Upon a ligand binding to its receptor, RAS is activated and triggers a phosphorylation cascade in which MAP3K phosphorylates MAP2K, and subsequently phosphorylates and activates MAPK. Once activated, MAPK translocates to the nucleus to regulate target gene expression.
Tumor progression locus 2 (Tpl2) is a gene encoding a serine/threonine kinase that plays an important role in inflammation, immunity, and cancer [8,9]. As a MAP3K, Tpl2 lies upstream of both the MAP2K MEK1/MEK2 and the MAPK ERK1/ERK2. Our prior research demonstrated that Tpl2 ablation in mice increases sensitivity to aberrant RAS signaling, resulting in heightened inflammation and tumorigenesis [10]. Additionally, the absence of Tpl2 leads to increased activation of several receptor-tyrosine kinase (RTK) bypass pathways, including those associated with the Epidermal Growth Factor Receptor (EGFR/ErbB) family [11,12]. The extent to which dysregulated EGFR signaling is responsible for the heightened tumorigenesis in Tpl2^−/−^ mice is unknown.
The ErbB family consists of four transmembrane receptors: ErbB1 (EGFR/HER1), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4) [13]. These RTKs function through ligand-induced homo- or heterodimerization, leading to activation of downstream signaling pathways that promote cell proliferation, survival, and migration. Overexpression of EGFR in high-risk or metastatic cutaneous squamous cell carcinoma (cSCC) is associated with poor prognosis, and emerging evidence suggests that EGFR inhibitors may offer potential benefit for patients with advanced disease [14,15,16]. Likewise, HER2 mutation or amplification is reported in non-melanoma skin cancers and HER2 loss significantly reduces DMBA/TPA-induced skin tumor formation [17,18].
MicroRNAs (miRNAs or MiRs) are short (~22 nucleotide) non-coding RNAs that are frequently dysregulated in cancer [19]. By binding to target mRNAs, microRNAs can suppress gene expression at the post-transcriptional level. In the skin, they serve as regulators in processes such as wound healing, epidermal differentiation, and the response to ultraviolet radiation [15]. Notably, they demonstrate dual functionality in cSCC, with some functioning as oncogenic miRNA (OncomiR) while others act as tumor suppressors [20]. Several microRNAs, including miR-7, miR-125, miR-21, and miR-205, regulate ErbB signaling by targeting expression of the receptors themselves or downstream components of their pathways [20,21].
In this study, we found heightened gene expression of ErbB receptors and ErbB-related microRNA 205 and 21 in keratinocytes from Tpl2^−/−^ mice. Additionally, immunohistochemical analysis of papillomas from Tpl2^−/−^ mice showed upregulation in EGFR, p-EGFR and HER2. Using a DMBA/TPA multi-stage chemical carcinogenesis protocol, we found that Tpl2^−/−^ mice developed significantly more tumors and experienced more rapid tumorigenesis compared to control mice. Mice fed the EGFR inhibitor Gefitinib reduced tumor burden by 88% in Tpl2^−/−^ mice, while the HER2 inhibitor Lapatinib achieved a 50% tumor reduction. The findings indicate that Tpl2 loss removes safeguards that normally limit ErbB-driven oncogenic processes, establishing the ErbB family as a key mechanistic link between Tpl2 deficiency and accelerated carcinogenesis.
2. Materials and Methods
2.1. In Vivo Tumor Experiment
Sixty male and female wild-type (Tpl2^+/+^) and knockout (Tpl2^−/−^) C57Bl/6 mice engineered as previously described were maintained at the American University (Washington, DC, USA) animal facility [22]. For two-stage chemical carcinogenesis studies, mice were randomized into groups (8–10 mice/group) matched for age, weight, and sex. Prior to initiation with 7,12-dimethylbenz(a)anthracene (DMBA; 100 μg/200 μL acetone), dorsal skin was shaved. Mice were promoted with twice weekly application of TPA painted on the skin (10 μg/200 μL acetone) for 20 weeks. At the time of promotion, mice were fed ad libitum an AIN-93G diet or AIN-93G diet containing 200 mg/kg Gefitinib, an EGFR tyrosine kinase inhibitor, or 200 mg/kg Lapatinib, an EGFR/HER2-targeted tyrosine kinase inhibitor. Control mice treated with only acetone, DMBA, or TPA were maintained for both genotypes. Mice were monitored daily for general appearance and tumor measurements and weights taken weekly. Tumor-bearing animals were individually housed to avoid injury to the tumor sites. Animals were euthanized 48 weeks after the date of initiation, or at an earlier time point if the animal was deemed moribund. At the study endpoint, portions of skin and tumors were either snap-frozen for DNA/RNA/protein isolation or formalin-fixed for Immunohistochemistry. Tumors underwent a histological examination in a blinded fashion by a certified pathologist to determine tumor type (Applied Pathology Systems; Shrewsbury, MA, USA).
2.2. Primary Keratinocyte Isolation and Treatment
Primary keratinocytes were isolated from Tpl2^−/−^ and wild-type Tpl2^+/+^ C57Bl/6 mice pups between 1 and 4 days old as previously described [23]. Keratinocytes were grown at 37 °C and 5% CO_2_ in keratinocyte growth media (Promocell; Heidelberg, Germany) containing hormone supplements, Penicillin-Streptomycin (10,000 U/mL) and low (0.06 mM) calcium. For in vitro drug studies, keratinocytes were treated with 1 μM Lapatinib or 1 μM Gefitinib (Selleck Chemicals; Houston, TX, USA) for 15 min (for assessing phosphorylated protein) or 24 h (for assessing total protein) prior to isolation.
2.3. RT-qPCR
RNA was isolated using Invitrogen’s PureLink RNA Mini Kit (Thermofisher Scientific; Waltham, MA, USA) and quantified with a NanoVue Plus Spectrophotometer (GE Healthcare (Chicago, IL, USA)). For ErbB receptor expression, cDNA was reverse transcribed from template RNA and qPCR was performed as previously described using specific forward and reverse primers for each gene target, with GAPDH as the housekeeping gene (Table 1) [24]. For microRNA gene expression, RNA was standardized to 100–125 ng/mL before reverse transcription using the Qiagen miRCURY LNA RT kit (Qiagen; Germantown, MD, USA). qPCR was performed with Qiagen’s miRCURY LNA SYBR Green PCR kit using miR-21a-3p (YP00205400) and miR-205-3p (YP02114511) sequences (Table 2). MiRNA abundance was determined by absolute quantification using synthetic oligonucleotides with serial dilutions of 10 mM to 0.001 mM (miR205) and 1 mM to 0.0001 mM (miR21). Pooled keratinocyte RNA from 4 to 6 mice was used, with experiments repeated a minimum of three times.
2.4. Immunohistochemistry
Immunohistochemistry was conducted as previously described [25]. Sections of skin, tumors, and SCCs from Tpl2^−/−^ and Tpl2^+/+^ mice, fixed in formalin and embedded in paraffin, were sectioned into 4 mm slices and stained with hematoxylin and eosin (H&E). Primary antibodies targeting the ErbB receptors along with anti-rabbit secondary antibodies (Cell Signaling #8339) were applied and used at concentrations of 1:200 to 1:600. Sections came from a minimum of three individual mice per treatment group. Representative areas were captured using 10× and 50× magnification.
High-magnification images were analyzed using the Fiji distribution of ImageJ2 (Version 2.17.0). Regions of interest (ROIs) encompassing areas of dense chromogen deposition were identified using empirically determined thresholds applied to pixel intensity, and then the total area within all ROIs was summed for each image. Identical thresholds were used for all images collected from papilloma and cSCC samples. Due to the weak immunolabeling of samples from normal skin, a different threshold was applied to these images in an attempt to minimize the inclusion of unlabeled cells that strongly took up the H&E stain; even with a more stringent threshold, the quantification values likely overrepresent the total area of chromogen deposition in the normal skin samples. Some images contained dark areas that were clearly not part of the tissue section (e.g., cellular debris from processing) or the structure of interest (e.g., a hair bulb near a papilloma or cSCC), but which registered as an ROI due to their average pixel intensity. When these areas were 100% identifiable and separable from the actual ROIs labeled with chromogen, they were manually cropped from the image before analysis. Quantification of IHC images is shown in Supplementary Materials.
2.5. Western Blotting
Keratinocyte protein was extracted using RIPA lysis buffer with protease/phosphatase inhibitors and quantified by BCA assay (Thermo Fisher; Waltham, MA, USA). 25 μg protein was electrophoresed using 4–12% Tris-Glycine gels, transferred to PVDF membranes, and blocked with 5% milk/BSA. Primary antibodies (EGFR, HER2, HER3, p-EGFR, β-actin, GAPDH; Cell Signaling Technologies, Danvers, MA, USA) were used at 1:500-1:1000, and secondary anti-rabbit HRP was applied at 1:2000. Bands were detected with West Dura substrate (Thermo Fisher), quantified using NIH ImageJ, and normalized to β-actin/GAPDH. Pooled protein from 4 to 6 mice was used with experiments repeated a minimum of 3 times.
2.6. Statistical Analysis
Data was tested for normality, model assumptions were checked, and the data were analyzed with SPSS software, version 28.01.01. For qPCR data, unpaired t-tests were used for statistical analysis. Tumor induction experiments were analyzed through two-way ANOVA with Tukey’s post hoc test. Significance for all analyses was assumed at a p-value of 0.05 or less. Significance values of p ≤ 0.05 are indicated in figures with a single asterisk (), p ≤ 0.01 with a double asterisk (), and p ≤ 0.001 with a triple asterisk ().
3. Results
3.1. ErbB Receptor Upregulation in Tpl2−/− Keratinocytes and Compensatory Response to Inhibitor Treatment
To assess whether the ErbB receptor tyrosine kinases EGFR, HER2, and HER3 were upregulated in Tpl2^−/−^ keratinocytes, qPCR was performed (Table 1, Figure 1). Tpl2^−/−^ keratinocytes exhibited a 2.4-fold increase in EGFR mRNA expression compared to Tpl2^+/+^ keratinocytes (Figure 1A). Similarly, Tpl2^−/−^ keratinocytes exhibited a 4.1-fold higher expression of HER2 mRNA and 4.4-fold higher expression of HER3 mRNA than Tpl2^+/+^ keratinocytes. Despite upregulation in gene expression, baseline protein levels in Tpl2^+/+^ and Tpl2^−/−^ keratinocytes were similar, suggesting post-transcriptional modulation (Figure 1B). Inhibition of p-EGFR using Gefitinib led to compensatory upregulation of EGFR family receptors in both Tpl2^+/+^ and Tpl2^−/−^ keratinocytes. In Tpl2^+/+^ cells, Gefitinib treatment increased EGFR, HER2, and HER3 protein expression by 1.7-, 1.9-, and 1.6-fold, respectively. Tpl2^−/−^ cells showed a similar pattern but with more pronounced HER2 upregulation (1.7-, 6.1-, and 1.8-fold increases for EGFR, HER2, and HER3, respectively). Lapatinib treatment produced comparable compensatory effects. Tpl2^+/+^ keratinocytes treated with Lapatinib demonstrated 1.9-, 1.9-, and 1.6-fold increases in EGFR, HER2, and HER3 protein, respectively, while Tpl2^−/−^ cells showed increases of 1.3-, 3.2-, and 1.2-fold, respectively. Given our previous findings that MET receptor (another RTK) is also activated in Tpl2^−/−^ keratinocytes, we also tested whether Capmatinib (a MET inhibitor) could increase ErbB receptors [11]. Capmatinib treatment increased p-EGFR, EGFR, and HER3 protein levels in Tpl2^+/+^ mice (1.7-,2.1-, and 1.4-fold, respectively), but not in Tpl2^−/−^ keratinocytes (Figure 1B).
3.2. miR205 and miR21 Are Upregulated in Tpl2−/− Keratinocytes
Given their established role in post-transcriptional regulation, microRNA analysis was performed to explore potential mechanisms underlying the observed gene-protein expression differences. Several miRNAs—namely miR-205, miR-7, miR-21, and miR-125—have been identified as key modulators of ErbB family members and play critical regulatory roles in cSCC (Figure 2A). Thus, we tested their expression in Tpl2^+/+^ and Tpl2^−/−^ keratinocytes. qPCR analysis revealed significant microRNA upregulation in Tpl2^−/−^ keratinocytes: miR-21 was increased 2.3-fold (p = 0.049) and miR-205 was increased 6.9-fold (p = 0.003) compared to Tpl2^+/+^ keratinocytes (Figure 2B,C). These results were consistent across a minimum of four independent biological replicates. miR-125 showed no genotype differences and miR-7 was barely detectable.
3.3. Pharmacological Inhibition of EGFR or HER2 Decreases Tumorigenesis in Tpl2−/− Mice
To investigate the role of EGFR/HER2 signaling in tumorigenesis, we used a two-stage chemical carcinogenesis model in Tpl2^+/+^ and Tpl2^−/−^ mice. Like our prior work, 100% of Tpl2^−/−^ mice developed skin tumors compared to only 30% of wildtype (Tpl2^+/+^) mice (Figure 3A). Tumor latency was significantly shorter in Tpl2^−/−^ mice (p < 0.0001), with 63% developing papillomas by week 14 compared to 0% of Tpl2^+/+^ mice. Tumor burden was also dramatically higher in Tpl2^−/−^ mice (54 tumors total) compared to Tpl2^+/+^ mice (4 tumors total) (p < 0.001; Figure 3B). Gefitinib and Lapatinib treatment reduced tumor burden in Tpl2^−/−^ mice by 88% and 50%, respectively, restoring tumor counts to Tpl2^+/+^ levels (Figure 3B,C). Histological analysis revealed a higher incidence of cSCCs in the Tpl2^−/−^ mice, with 4 observed compared to only 1 in the Tpl2^+/+^ mice. Notably, treatment with either Gefitinib or Lapatinib reduced the number of cSCCs in the Tpl2^−/−^ mice to 1 in each treatment group (Table 3).
3.4. Papillomas from Tpl2−/− Mice Have Increased Expression of EGFR, p-EGFR and HER2
Skin, papillomas, and cSCCs were obtained from Tpl2^+/+^ and Tpl2^−/−^ mice and were stained for EGFR, p-EGFR, HER2, p-HER2, HER3 or p-HER3. Tpl2^−/−^ papillomas showed increased expression of EGFR, p-EGFR, and HER2 compared to Tpl2^+/+^ mice (Figure 4A,B). cSCCs had lower expression of all ErbB-related receptors compared to papillomas, and similar expression patterns between genotypes, suggesting that upregulation of RTKs in Tpl2^−/−^ papillomas may be required for malignant transformation to cSCCs. For IHC analysis, a minimum of three tissue sections from three individual mice were examined. Since only one cSCC developed in Tpl2^+/+^ mice in this study, two additional sections from Tpl2^+/+^ cSCCs obtained from a parallel study were included to ensure reproducibility.
4. Discussion
Growing evidence suggests that cSCC development and progression may arise from aberrant MAPK signaling pathways. [5,26] While MAPK inhibitors show promise as a cancer therapeutic strategy, their clinical application is complicated by the development of secondary malignancies [7,27]. This paradox is particularly evident in BRAF-mutant melanoma, where these inhibitors effectively target critical oncogenic pathways yet simultaneously increase the risk of cSCC [27]. This effect is also evident following Tpl2 (MAP3K) loss, as we have previously reported that Tpl2 ablation results in a significantly higher number of papillomas and cSCCs [10,11].
Here we demonstrate that MAP3K signal disruption can upregulate compensatory ErbB pathways that maintain pro-tumorigenic signaling. Tpl2 ablation leads to increased gene expression of EGFR, HER2, and HER3 in Tpl2^−/−^ keratinocytes and increased EGFR, p-EGFR, and HER2 protein levels in Tpl2^−/−^ papillomas. EGFR signaling drives proliferation and pro-inflammatory cytokine/chemokine upregulation, promoting tumorigenesis and therapy resistance [13]. This is consistent with our previous findings of increased tumorigenesis and inflammation in Tpl2^−/−^ mice [10,11,25].
Upon administration of Gefitinib or Lapatinib to keratinocytes, HER2 and HER3 signaling pathways become upregulated, indicating the activation of compensatory bypass mechanisms. This adaptive response demonstrates the tumor’s ability to circumvent targeted therapeutic blockades by redirecting cellular signaling through alternative receptor networks. While this occurred in both genotypes, the upregulation in HER2 signaling following Gefitinib or Lapatinib treatment was more pronounced in Tpl2^−/−^ keratinocytes.
MicroRNAs are key regulators in cSCC, particularly through their influence on ErbB signaling. Among the miRNAs implicated, miR-205, miR-7, miR-21, and miR-125 play prominent roles in modulating ErbB receptor activity [20]. miR-7 and miR-125 typically function as tumor suppressors by targeting EGFR or HER2/3, respectively, or their downstream signaling components [28]. In contrast, miR-21 is frequently overexpressed in cSCC [20,29,30]. miR-205 demonstrates context-specific behavior, acting as either a tumor suppressor or promoter depending on the tissue type. [20] However, in cSCC, MiR-205 is oncogenic, as it is reported to be more frequently expressed in cSCCs with aggressive traits, including association with local recurrence, promotion of keratinocyte migration, and correlation with clinical indicators of poor patient outcomes [29,31,32]. Consistent with these findings, we observed significantly elevated levels of miR-205 and miR-21 in keratinocytes derived from Tpl2^−/−^ mice. Although we observed upregulation of two miRNAs, we acknowledge this does not provide direct functional evidence of their ability to inhibit ErbB signaling in our model. Future studies will employ miRNA overexpression/knockdown approaches and investigate additional post-transcriptional mechanisms to establish clearer causal relationships and determine which post-transcriptional regulatory mechanisms are responsible for the observed gene-protein discrepancies.
To determine whether ErbB signaling drives the increased tumorigenesis observed in Tpl2^−/−^ mice, we evaluated the effects of EGFR or HER2 inhibition on tumor development and cSCC progression in our model. Tpl2^−/−^ mice developed 12-fold more papillomas and 4-fold more cSCCs than Tpl2^+/+^ animals. However, while substantial numbers of papillomas formed in Tpl2^−/−^ mice, only four cSCCs developed in untreated Tpl2^−/−^ mice compared to one in Tpl2^+/+^ mice and one in drug-treated Tpl2^−/−^ mice. These limited cSCC counts constrained our molecular characterization of the malignant lesions. Future investigations will necessitate larger animal cohorts and potentially longer observation periods to obtain adequate cSCC numbers for comprehensive analysis of therapeutic effects on malignant progression.
Treatment with the EGFR inhibitor Gefitinib or EGFR/HER2 inhibitor Lapatinib resulted in a decrease in the number of papillomas by 88% and 50%, respectively, and restored the number of cSCCs back to Tpl2^+/+^ levels. The superior efficacy of Gefitinib compared to Lapatinib suggests that EGFR signaling may be the predominant driver of tumorigenesis in this model, consistent with our observation that p-EGFR was the most strongly upregulated phospho-receptor in Tpl2^−/−^ papillomas. Our research extends prior studies showing clinical efficacy of gefitinib and lapatinib in cSCC treatment and provides mechanistic evidence supporting EGFR as a primary therapeutic target in Tpl2-deficient tumors [33,34].
5. Conclusions
In summary, we propose that compensatory upregulation of ErbB signaling plays a significant role in driving cSCC development when Tpl2 is absent. These findings suggest that ErbB inhibition warrants further investigation as a potentially promising and targeted therapeutic intervention for this specific subset of cSCC tumors characterized by disruptions in MAPK signaling.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Eggermont C.J. Eggermont A.M.M. Shifting landscape in skin cancer incidence: The rising tide of cutaneous squamous cell carcinoma and potential implications for prevention Br. J. Dermatol.202419046046110.1093/bjd/ljad 48038099564 · doi ↗ · pubmed ↗
- 2Urban K. Mehrmal S. Uppal P. Giesey R.L. Delost G.R. The global burden of skin cancer: A longitudinal analysis from the Global Burden of Disease Study, 1990–2017 JAAD Int.202129810810.1016/j.jdin.2020.10.01334409358 PMC 8362234 · doi ↗ · pubmed ↗
- 3Jiang R. Fritz M. Que S.K.T. Cutaneous Squamous Cell Carcinoma: An Updated Review Cancers 202416180010.3390/cancers 1610180038791879 PMC 11119634 · doi ↗ · pubmed ↗
- 4Corchado-Cobos R. García-Sancha N. González-Sarmiento R. Pérez-Losada J. Cañueto J. Cutaneous Squamous Cell Carcinoma: From Biology to Therapy Int. J. Mol. Sci.202021295610.3390/ijms 2108295632331425 PMC 7216042 · doi ↗ · pubmed ↗
- 5Chang D. Shain A.H. The landscape of driver mutations in cutaneous squamous cell carcinomanpj Genom. Med.202166110.1038/s 41525-021-00226-434272401 PMC 8285521 · doi ↗ · pubmed ↗
- 6Guo Y.J. Pan W.W. Liu S.B. Shen Z.F. Xu Y. Hu L.L. ERK/MAPK signalling pathway and tumorigenesis Exp. Ther. Med.2020191997200710.3892/etm.2020.845432104259 PMC 7027163 · doi ↗ · pubmed ↗
- 7Braicu C. Buse M. Busuioc C. Drula R. Gulei D. Raduly L. Rusu A. Irimie A. Atanasov A.G. Slaby O. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer Cancers 201911161810.3390/cancers 1110161831652660 PMC 6827047 · doi ↗ · pubmed ↗
- 8Gantke T. Sriskantharajah S. Ley S.C. Regulation and function of TPL-2, an IκB kinase-regulated MAP kinase kinase kinase Cell Res.20112113114510.1038/cr.2010.17321135874 PMC 3193413 · doi ↗ · pubmed ↗