Identification of Fecal Microbiota and Related Metabolites Associated with Feed Efficiency in DLY Pigs
Zhicheng Zhang, Kuirong Chen, Shuai Zhang, Yiyun He, Guofeng Lei, Yunxiang Zhao, Jing Liang

TL;DR
This study identifies gut microbes and metabolites linked to better feed efficiency in pigs, which could help improve breeding and reduce costs.
Contribution
The study identifies specific gut microbes and metabolites associated with feed efficiency in pigs, offering new insights for targeted breeding strategies.
Findings
Pigs with lower feed conversion ratios had higher abundances of beneficial bacteria like Ruminococcus, Prevotella, Akkermansia, and Eubacterium.
Metabolite differences in steroid hormone biosynthesis and bile acid metabolism were observed between high and low feed efficiency pigs.
Certain gut microbes correlated with bile acid metabolites and steroid hormone synthesis, suggesting their role in feed efficiency.
Abstract
Improving feed efficiency (FE) is crucial for modern pig breeding due to its significant economic and environmental impacts. Fecal microbiota play a critical role in this by synthesizing various beneficial substances that enhance FE. This study utilized 16S rRNA gene sequencing and LC-MS to analyze microbial and metabolic differences between pigs with high and low feed conversion ratios (FCR). Results indicated that pigs with lower FCR had an increased abundance of beneficial SCFA-producing bacteria, while higher-FCR pigs exhibited greater levels of pathogenic taxa. These findings highlight microbial and metabolic contributions to feed efficiency and provide insights into targeted breeding strategies. Improving feed efficiency (FE) is essential for enhancing productivity, reducing production costs, and minimizing environmental impacts in the swine industry. Fecal microbiota and their…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
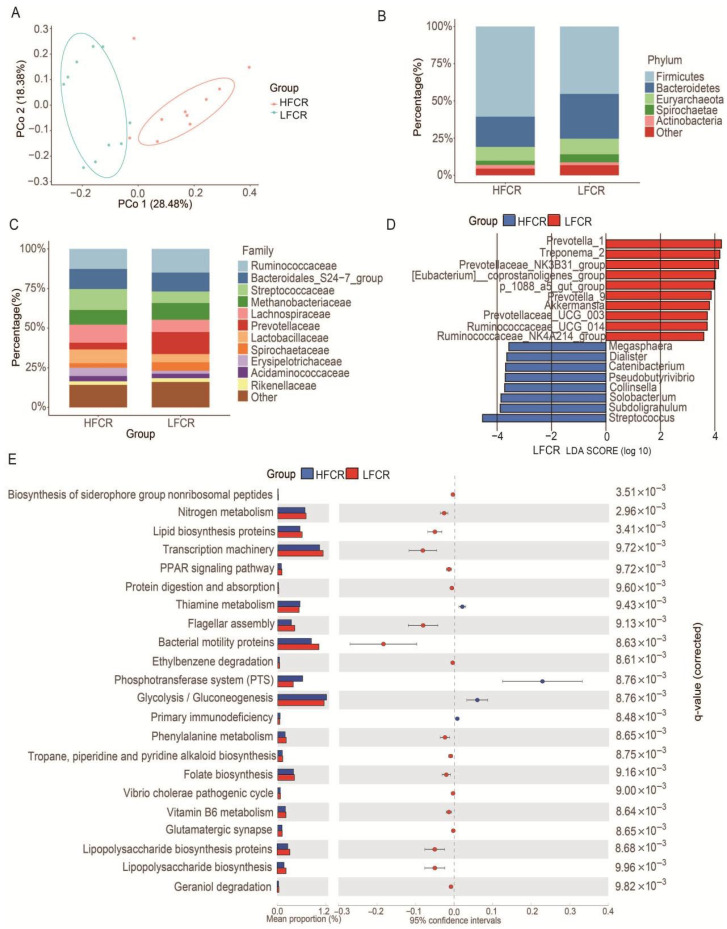 Figure 1
Figure 1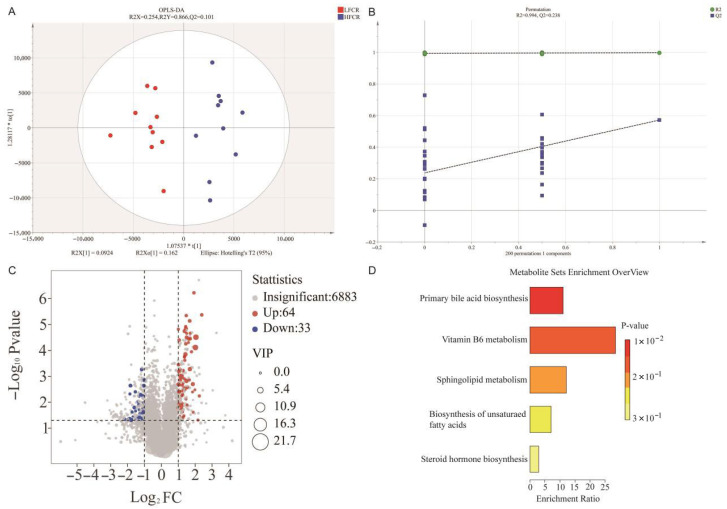 Figure 2
Figure 2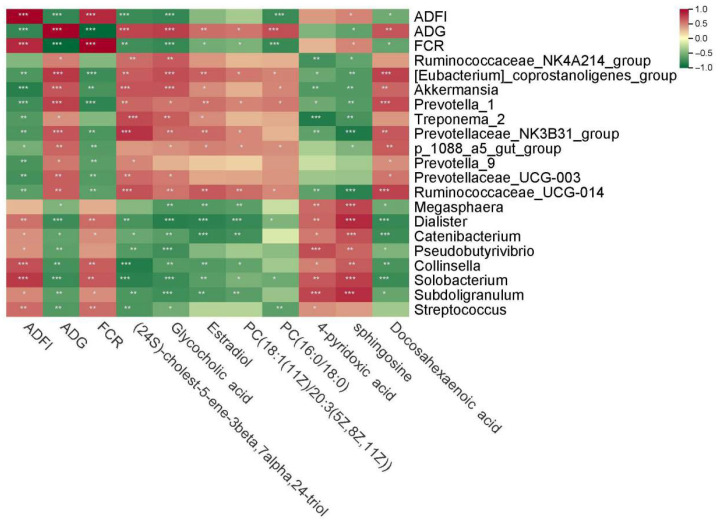 Figure 3
Figure 3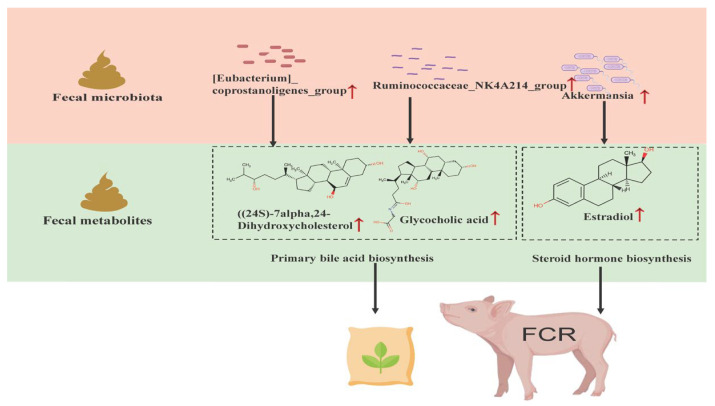 Figure 4
Figure 4- —National Key R&D Program of China
- —Guangxi “open competition” Technology Project, research and application of pig genome selection breeding technology
- —Guangxi Pig Industry Innovation Team Building Program of National Modern Agricultural Industrial Technology System
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGut microbiota and health · Clostridium difficile and Clostridium perfringens research · Animal Nutrition and Physiology
1. Introduction
Pork constitutes the principal meat consumed worldwide and is a significant source of high-quality protein and key micronutrients [1]. As the global population expands and household incomes continue to rise, worldwide pork consumption is projected to double by 2050 [2]. Therefore, finding ways to enhance pork production and fulfill human needs has become a pressing challenge in modern animal agriculture.
Feed costs account for approximately 60% of the total production expenses in pig farming, making FE a critical determinant of economic sustainability and profitability [3]. The main indices for evaluating FE are residual feed intake (RFI) and FCR [4]. RFI represents the difference between actual feed intake and expected feed intake [5], while FCR is defined as the ratio of average daily feed intake (ADFI) to average daily gain (ADG) [6]. The adoption of automated feeding systems has greatly improved the precision and convenience of monitoring these metrics, thereby facilitating data-driven selection in pig breeding programs [7].
FE is influenced by a combination of genetic, physiological, and environmental factors, with increasing evidence highlighting the role of fecal microbiota in modulating feed utilization [8]. These microbial communities are capable of fermenting complex carbohydrates, such as cellulose and resistant starches, which are otherwise indigestible by the host. This fermentation process produces short-chain fatty acids (SCFAs), which serve as energy sources for the host [9]. Previous studies have reported microbial taxa associated with divergent feed efficiency, such as the enrichment of Lachnospiraceae and Prevotellaceae, together with Streptococcus (notably Streptococcus gallolyticus subsp. gallolyticus), in high-FE DLY pigs [10], and enrichment of Prevotella CAG:604 in the cecum of pigs with high FCR (HFCR) [11]. Similarly, McCormack et al. found that an augmentation of specific fecal microbiota in pigs with low RFI was associated with improved health and FE [12]. These findings underscore the importance of fecal microbiota in shaping host metabolic performance and feed conversion.
Although fecal metabolomics offers valuable insights into microbial metabolic activity and host nutrient utilization, its application in feed efficiency studies remains limited [13,14]. Fecal metabolites, as the end products of host-microbe interactions, reflect both microbial composition and gut physiological processes. They serve as functional biomarkers for investigating the links between microbiota metabolism and host phenotypes [15]. Metabolomic analysis enables the identification of differential metabolites, offering a deeper understanding of metabolic pathways involved in nutrient absorption and energy balance. This integrative approach may support the development of targeted strategies to enhance FE in pig production.
Despite growing interest, the combined effects of fecal microbiota composition and fecal metabolites on FE remain inadequately understood. In this study, we employed 16S rRNA gene sequencing and liquid chromatography–mass spectrometry (LC/MS) to profile the fecal microbiota and metabolites in pigs with contrasting FCR. By integrating microbiome and metabolome data, we aimed to uncover key microbial and metabolic signatures associated with feed efficiency, thereby providing new targets for precision breeding and nutritional interventions in swine production.
2. Materials and Methods
2.1. Animal Management, Phenotypic Data Collection, and Sampling
A total of 384 Duroc × (Landrace × Yorkshire) (DLY) pigs were raised under uniform conditions on a commercial farm in Nanning, Guangxi, China. Individual feed intake and body weight were monitored from 90 to 165 days of age using an automatic feeding system (Nedap N.V., Groenlo, The Netherlands). ADFI, ADG, and FCR (FCR = ADFI/ADG) were calculated. Based on FCR values, 10 pigs with the highest FCR values as the HFCR group, and 10 pigs with the lowest FCR values as the LFCR group, were selected for further study. All pigs received the same diet ad libitum, had free access to water, remained clinically healthy, and were not treated with antibiotics during the trial. The ingredient composition and nutrient levels of the experimental diet are provided in Supplementary Table S1. At 170 days of age, fresh fecal samples were collected from all 384 pigs on the same day, immediately frozen in liquid nitrogen, transported on dry ice, and stored at −80 °C until analysis. Detailed growth performance for the HFCR and LFCR groups is provided in Supplementary Table S2, and the growth data for all 384 pigs are provided in Supplementary Table S3.
2.2. Fecal Microbiota Analysis
Total genomic DNA was extracted from fecal samples using the QIAamp Fast DNA Fecal Kit (Qiagen, Hilden, Germany). Negative controls were included during DNA extraction and subsequent PCR amplification to monitor for contamination. DNA concentration and integrity were assessed via Qubit fluorometry and 1% agarose gel electrophoresis. The V3–V4 region of the 16S rRNA gene was amplified using primers 341F (5′-CCTACGGGNGGWGCAG-3′) and 806R (5′-GACTACHVGGGTWTCTAAT-3′) with barcodes. PCR products with expected size and concentration (≥2 ng/μL) were purified and used to construct sequencing libraries. Libraries were quantified (≥4 nM), pooled, and sequenced on an Illumina MiSeq platform using the PE250 strategy. Amplification sub-process analysis: Bioinformatics analysis of the amplicons was conducted using EasyAmplicon (v1.0). After quality control of the original Illumina fastq file (with an error rate < 1%), redundancy was removed using VSEARCH with the parameter min_Unique_Size set to 59 [16]. Usearch (v11) was used to perform a 100% similarity comparison of ASVs, followed by the removal of plastids and non-bacterial sequences using the sintax_Cutoff parameter set to 0.1. Low-abundance ASVs were filtered using Vegan (v2.6.4) packages, and all samples were normalized to 99,888 sequences to determine ASV abundance. Species annotation was performed using the SILVA database (v138), and the results were used for amplicon analysis [17]. Alpha diversity indices (Richness, Chao1, ACE, Shannon, Simpson) were calculated using the Vegan package. Beta diversity was analyzed by Bray–Curtis distance-based Principal Coordinate Analysis (PCoA). Taxonomic composition was visualized at the phylum and family levels, and LEfSe (http://www.bic.ac.cn/BIC/, accessed on 15 March 2024) was used to identify differentially abundant genera (LDA > 3).
2.3. Fecal Metabolomics Analysis
Take the sample from the −80 °C refrigerator and thaw it on ice (all subsequent operations were performed on ice). Weigh 20 mg (±1 mg) of feces and transfer it to the appropriately labeled centrifuge tube. Add 400 µL of 70% methanol-water internal standard extraction solution and vortex for 3 min. If the sample is not dispersed, add steel balls and vortex for an additional 3 min. Sonicate in an ice-water bath for 10 min, then remove the sample and vortex for 1 min. Next, place the sample in a −20 °C freezer for 30 min. Centrifuge at 12,000× g for 10 min at 4 °C, and transfer 300 μL of the supernatant to a fresh microcentrifuge tube. Then, centrifuge at 12,000× g for 3 min and extract 200 µL of the supernatant for subsequent machine analysis [18]. Supernatants were used for LC-MS analysis. Samples were analyzed using a Triple TOF-6600 mass spectrometer (AB Sciex, Framingham, MA, USA) in both positive and negative ion modes. Chromatographic separation was performed on an LC20 UHPLC system (Shimadzu, Kyoto, Japan) with a Waters ACQUITY UPLC HSS T3 column. Mobile phases consisted of water with 0.1% formic acid (A) and acetonitrile with 0.1% formic acid (B).
Raw data were converted to mzXML using ProteoWizard and processed with XCMS for peak detection and alignment. Peaks with >50% missing values were excluded. After calibration and filtering, a metabolic spectrum table was compiled by querying the proprietary database (Wuhan Maiwei Metabolic Biotechnology Co., Ltd., Wuhan, China). Quality control was assessed using total ion current (TIC) overlays from pooled QC samples (Supplementary Figure S1). Normalized data were subjected to orthogonal partial least squares discriminant analysis (OPLS-DA) in SIMCA-P (version 14.1). Differential metabolites were identified using VIP > 1, p < 0.05, and FC > 2 or < 0.5. Annotated metabolites were mapped to the Human Metabolome Database (HMDB) and analyzed using MetaboAnalyst 5.0 and KEGG for pathway enrichment (Supplementary Table S4).
2.4. Statistical Analysis
Phenotypic traits were analyzed using SPSS version 26.0. Results are expressed as mean ± standard deviation (SD), and 95% confidence intervals (CI) are also reported to indicate the precision of estimates. Differences in alpha diversity and metabolite levels between groups were assessed using two-sided independent-samples t-tests with significance set at p < 0.05. Pairwise Spearman correlations (|ρ| > 0.6, p < 0.05) were computed among phenotypic traits, differential taxa, and metabolites to construct association networks.
3. Results
3.1. Phenotypic Differences Between HFCR and LFCR Pigs
Significant differences in growth traits were observed between HFCR and LFCR groups. Pigs in the LFCR group exhibited a significantly higher ADG, along with lower ADFI and FCR compared to those in the HFCR group (p < 0.001; Table 1).
3.2. Fecal Microbiota Composition Differences Between HFCR and LFCR Pigs
Alpha diversity metrics (Richness, Chao1, ACE) were significantly higher in LFCR pigs compared to HFCR pigs (p < 0.01), indicating greater microbial abundance. The Shannon index was also higher in LFCR pigs, though the difference was marginally significant (p = 0.05; Table 2).
Principal Coordinate Analysis (PCoA) based on Bray–Curtis distances showed distinct microbial community structures between groups (PC1 = 28.48%, PC2 = 18.38%; Figure 1A). At the phylum level, Firmicutes and Bacteroidetes dominated both groups, with LFCR pigs showing higher Bacteroidetes and lower Firmicutes abundance (Figure 1B). At the family level, the LFCR group shows higher abundances of Ruminococcaceae and Prevotellaceae and lower abundances of Streptococcaceae and Lachnospiraceae than the HFCR group (Figure 1C). For the genus-level microbiota, genera with an average relative abundance greater than 0.5% were selected for LefSe analysis (LDA > 3; Figure 1D). LFCR-enriched genera included Prevotella_1, Treponema_2, Prevotelaceae_NK3B31_group, [Eubacterium]_coprostanogenes_group, p_1088_a5_gut_group, Prevotella_9, Akkermansia, Prevotelaceae_UCG-003, Ruminococcaceae_UCG-014, Ruminococcaceae_NK4A214_group. HFCR-enriched genera included Megaspaera, Dialist, Catenibacterium, Pseudobotyrivibrio, Collinsella, Solobacterium, Subdoligranulum, and Streptococcus. PICRUSt-based functional predictions revealed enhanced nitrogen metabolism and lipid biosynthesis potential in the LFCR group, whereas HFCR pigs had stronger functions in thiamine metabolism and phosphate transport systems (Figure 1E).
3.3. Fecal Metabolite Differences Between HFCR and LFCR Pigs
OPLS-DA modeling revealed clear separation between groups (R2X = 0.254, R2Y = 0.866, Q2 = 0.101; Figure 2A), with permutation testing confirming model robustness (Q2 = −0.376; Figure 2B). In total, 97 differential metabolites were identified (VIP > 1, p < 0.05, FC > 2 or < 0.5), including 64 upregulated and 33 downregulated in LFCR pigs (Figure 2C). KEGG pathway enrichment analysis indicated that differential metabolites were mainly involved in primary bile acid biosynthesis, vitamin B6 metabolism, sphingolipid metabolism, unsaturated fatty acid metabolism, and steroid hormone synthesis (Figure 2D). Representative metabolites are summarized in Table 3.
3.4. Correlation Analysis Among Phenotypes, Microbiota, and Metabolites
Spearman correlation analysis (correlation coefficient > 0.6, p < 0.05) identified relationships among phenotypic traits, differential microbes, and metabolites (Figure 3). Ruminococcus and Prevotella were negatively correlated with FCR, while Collinsella, Solobacterium, Subdoligranulum and Catenibacterium showed significant positive correlations with FCR. Additionally, the [Eubacterium]_coprostanogenes_group and Ruminococcaceae_NK4A214_group were positively correlated with bile acid-related metabolites, while Akkermansia was positively correlated with estradiol.
4. Discussion
4.1. Differences in Fecal Microbiota Between High and Low Feed Conversion Efficiency Pigs
Alpha diversity indices are widely used to evaluate microbial richness and evenness. Previous studies have linked higher microbial diversity to improved pig growth performance. For example, Tan et al. reported significantly higher alpha diversity in the fecal microbiota of LFCR pigs, a finding consistent with our results [19]. Similarly, Metzler et al. observed that pigs with low RFI exhibited greater cecal microbial diversity [20]. In our study, LFCR pigs showed significantly elevated values for Richness, Chao1, and ACE indices compared to HFCR pigs, suggesting a more diverse and possibly more metabolically beneficial gut microbial community that may support enhanced nutrient utilization and overall health.
Gut microbes play a critical role in fermenting undigested dietary components, producing metabolites such as SCFAs and bile acids that exert various physiological effects. Notably, members of Prevotellaceae [21] and Ruminococcaceae [22] are key fiber-degrading taxa that contribute significantly to SCFA production. Prevotella_1 specializes in breaking down hemicellulose or xylan, thereby supplying energy [23]. Similarly, Prevotellaceae_UCG-003 has been noted for its role in intestinal regulation and its capacity to break down fibrous polysaccharides into SCFAs. Prevotellaceae_NK3B31_group is associated with increased acetic acid, propionic acid, and total SCFAs, particularly in weaned piglets [24]. Moreover, the Ruminococcaceae_NK4A214_group demonstrates increased abundance in the rumen of low RFI cattle [25], a finding paralleling our own observations. Research indicates that Ruminococcaceae UCG-014 has the capability to produce butyric acid [26]. Additionally, [Eubacterium]_coprostanogenes_group contributes to SCFA production [27]. Akkermansia, recognized as a significant fecal biomarker linked with weight gain [28], shows that reduced Akkermansia abundance is directly associated with increased adiposity, insulin resistance, and dyslipidemia in obese mice [29]. Studies also highlight Akkermansia’s mucin-degrading capability and its role as an SCFA producer [30]. The enrichment of these SCFA-producing bacteria in LFCR pigs may enhance nutrient absorption and gut health, ultimately improving feed conversion efficiency.
Interestingly, some SCFA-producing taxa were also enriched in HFCR pigs. For example, Prevotella_1 and Prevotellaceae_UCG_003 were reported by Xue et al. to be more abundant in high RFI sheep [31]. Pseudobutyrivibrio, enriched in the distal colon, is positively associated with free fatty acid levels [32]. Subdoligranulum and Megaspaera, which convert lactate to SCFAs, were significantly enriched in the cecum of HFCR Landrace pigs [33]. However, Subdoligranulum has also been positively associated with depressive phenotypes in humans [34], suggesting that the physiological effects of SCFAs may be context-dependent and require further clarification.
Moreover, several bacteria enriched in HFCR pigs have been associated with adverse health outcomes. Collinsella has been linked to obesity, atherosclerosis [35], and hypercholesterolemia [36]. Lahti reported a significant positive correlation between Collins bacteria and TG (triglycerides) levels [37]. Solobacterium, part of the Erysipelotrichaceae family, is implicated in obesity, inflammatory bowel disease (IBD) [38], and colorectal cancer [39]. Streptococcus, a common swine pathogen and potential zoonotic agent, is a reservoir for antibiotic resistance genes [40]. The enrichment of these potentially pathogenic or dysbiotic bacteria in HFCR pigs may impair gut health and contribute to reduced feed efficiency.
4.2. Differential Fecal Metabolites Between High and Low Feed Conversion Efficiency Pigs
Bile acids are essential for lipid digestion, nutrient absorption, and energy metabolism. Their synthesis and transformation are closely linked to gut microbial activity and feed utilization efficiency in livestock [41]. In our study, the LFCR group exhibited significantly higher levels of bile acid-related metabolites, including (24S)Cholest-5-ene-3β,7α,24-triol and glycocholic acid. The former is a bile alcohol that arises during bile acid metabolism, while the latter is a conjugated bile acid with antibacterial properties. Bile alcohol can be seen as an intermediate and byproduct of the normal pathway in bile acid biosynthesis, playing a crucial role in food digestion and nutrient absorption [13]. Primary bile acids combine with glycine and taurine to form conjugated bile acids. Binding bile acids are influenced by microorganisms in the intestine to remove binding and exert their effects. Glycine bile acid is a conjugated bile acid with certain antibacterial properties [42]. The metabolic products associated with bile acids were notably higher in the LFCR group compared to the HFCR group, suggesting enhanced bile acid biosynthesis and transformation in pigs. This enhancement likely boosts the absorption of dietary fats and fat-soluble vitamins, leading to increased energy assimilation.
Unsaturated fatty acids, particularly omega-3 polyunsaturated fatty acids (PUFAs) like docosahexaenoic acid (DHA), possess multiple metabolic benefits, including anti-inflammatory effects [43], lipid-lowering activity [44], and cardiovascular protection [45]. PUFAs have also been shown to modulate fecal microbiota, notably increasing Akkermansia abundance, improving intestinal barrier integrity, and enhancing mucosal immunity [46]. Steroid hormones can affect the production of growth hormone by regulating endocrine factors related to growth [47]. As a steroid hormone, estrogen exerts pivotal regulatory control over reproductive tract and mammary gland development in sows [48]. The elevated levels of DHA and estradiol observed in LFCR pigs may contribute to superior growth performance and feed efficiency.
4.3. Integrated Microbiome-Metabolome Analysis Reveals Determinants of Feed Efficiency
Integrated correlation analysis revealed significant positive associations between bile acid–related metabolites (e.g., (24S)-Cholest-5-ene-3β,7α,24-triol, Glycocholic acid) and the bacterial taxa [Eubacterium]_coprostanogenes_group and Ruminococcaceae_NK4A214_group. Gut microbes are known to mediate bile acid dehydroxylation and transformation, converting primary to secondary bile acids [49]. Bile acids play a crucial role in bile acid metabolism and promote lipid digestion and absorption, regulate cholesterol metabolism, and stimulate bile secretion [50]. Taxa such as Bacteroides, Clostridium, Escherichia, and Eubacterium have demonstrated dehydroxylation effects [51]. Notably, [Eubacterium]_coprostanogenes_group has been reported to produce sterol derivatives [27], supporting its role in bile acid metabolism. Similarly, Ruminococcaceae_NK4A214_group has been shown to increase bile acid content when transplanted into mice, further supporting its contribution to improved fat and vitamin absorption, thereby enhancing feed conversion efficiency [52].
Furthermore, a strong positive correlation was observed between Akkermansia and estradiol. Prior studies indicate that Akkermansia supplementation can reduce obesity, inflammation, and insulin resistance [29]. Estradiol treatment has also been shown to increase the abundance of Akkermansia in ovariectomized mice [53]. In women, oral administration of heat-killed Akkermansia led to significant reductions in fat mass [54]. These findings suggest that Akkermansia may act synergistically with estradiol to support growth and improve feed efficiency in pigs (Figure 4).
4.4. Limitations and Future Directions
This study is observational and cross-sectional with a relatively small sample size for the fecal microbiota and metabolomics analyses; therefore, the findings should be interpreted as associations rather than causal effects. Although animals were matched for environment, genetic background, and sex, residual confounding cannot be fully excluded. To improve generalizability, subsequent studies should expand the cohort and include independent populations. In addition, we did not perform functional validation for the specific microbes and metabolites identified here. Future work will incorporate fecal microbiota transplantation (FMT) to probe microbial functions and controlled pig feeding trials to test the functional roles of candidate metabolites.
5. Conclusions
This study revealed that pigs with LFCR harbor a more diverse fecal microbiota enriched in SCFA-producing bacteria, while HFCR pigs show increased levels of potentially pathogenic microbes. Metabolomic analysis identified differential enrichment of bile acid and steroid hormone-related metabolites. Notably, Ruminococcaceae_NK4A214_group and [Eubacterium]_coprostanogenes_group were associated with bile acid metabolism, and Akkermansia correlated with estradiol levels. Together, our results indicate that fecal microbiota and their metabolites are associated with feed efficiency. While causality remains to be established, the identified microbial and metabolic markers suggest actionable targets for precision breeding, probiotic supplementation, and dietary interventions in swine.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Drewnowski A. Perspective: The Place of Pork Meat in Sustainable Healthy Diets Adv. Nutr.20241510021310.1016/j.advnut.2024.10021338508316 PMC 11035016 · doi ↗ · pubmed ↗
- 2Vargas D.A. Blandon S.E. Sarasty O. Osorio-Doblado A.M. Miller M.F. Echeverry A. Shelf-Life Evaluation of Pork Loins as Influenced by the Application of Different Antimicrobial Interventions Foods 202211346410.3390/foods 1121346436360077 PMC 9654175 · doi ↗ · pubmed ↗
- 3Si J. Feng L. Gao J. Huang Y. Zhang G. Mo J. Zhu S. Qi W. Liang J. Lan G. Evaluating the association between feed efficiency and the fecal microbiota of early-life Duroc pigs using 16S r RNA sequencing AMB Express.20201011510.1186/s 13568-020-01050-232562009 PMC 7305293 · doi ↗ · pubmed ↗
- 4Touitou F. Tortereau F. Bret L. Marty-Gasset N. Marcon D. Meynadier A. Evaluation of the Links between Lamb Feed Efficiency and Rumen and Plasma Metabolomic Data Metabolites 20221230410.3390/metabo 1204030435448491 PMC 9029153 · doi ↗ · pubmed ↗
- 5Yu W. Lu Y. Shen Y. Liu J. Gong S. Yu F. Huang Z. Zou W. Zhou M. Luo X. Exploring the Intestinal Microbiota and Metabolome Profiles Associated with Feed Efficiency in Pacific Abalone (Haliotis discus hannai)Front. Microbiol.20221385246010.3389/fmicb.2022.85246035369429 PMC 8969561 · doi ↗ · pubmed ↗
- 6Homma C. Hirose K. Ito T. Kamikawa M. Toma S. Nikaido S. Satoh M. Uemoto Y. Estimation of genetic parameter for feed efficiency and resilience traits in three pig breeds Animal 20211510038410.1016/j.animal.2021.10038434757251 · doi ↗ · pubmed ↗
- 7Gilbert H. Billon Y. Brossard L. Faure J. Gatellier P. Gondret F. Labussière E. Lebret B. Lefaucheur L. Le Floch N. Review: Divergent selection for residual feed intake in the growing pig Animal 2017111427143910.1017/S 175173111600286 X 28118862 PMC 5561440 · doi ↗ · pubmed ↗
- 8Zhang J. Jiang Q. Du Z. Geng Y. Hu Y. Tong Q. Song Y. Zhang H.Y. Yan X. Feng Z. Knowledge graph-derived feed efficiency analysis via pig gut microbiota Sci. Rep.2024141393910.1038/s 41598-024-64835-638886444 PMC 11182767 · doi ↗ · pubmed ↗