Cardiomyopathies: Temporal Review and Genetic Determination
Gaetano Thiene, Stefania Rizzo, Cristina Basso

TL;DR
This paper reviews the history and genetic causes of cardiomyopathies, including newly recognized electrical dysfunction types.
Contribution
The paper expands the definition of cardiomyopathies to include electrical dysfunction without structural changes, emphasizing genetic factors.
Findings
Cardiomyopathies now include electrical disorders like channellopathies.
Genetic background and gene therapy are key areas of focus in modern cardiomyopathy research.
Abstract
Cardiomyopathies are a heterogeneous group of diseases of the myocardium associated with dysfunction, with or without a structural substrate. They are frequently genetically determined. The dysfunction may be mechanical, both of the systole and diastole, or electrical, including arrhythmias or conduction disorders. Originally, only dilated, hypertrophic, restrictive–obliterative and arrhythmogenic dysfunctions were considered cardiomyopathies. Nowadays, since dysfunction can also be electric, disorders affected by electrical dysfunction without a structural substrate can be regarded as cardiomyopathies as well. This is the case of channellopathies and ryanodine receptors. This paper is a review of the history of cardiomyopathies, including the issues of their classification and nomination, genetic background and gene therapy.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1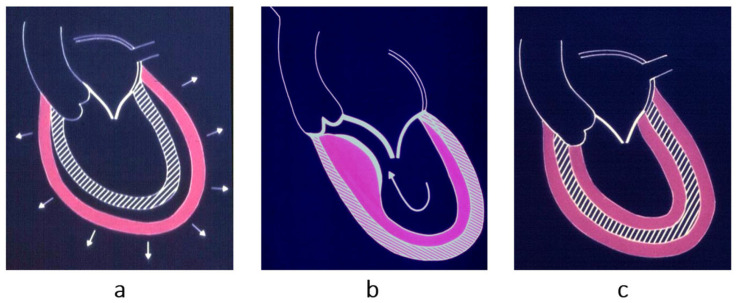 Figure 2
Figure 2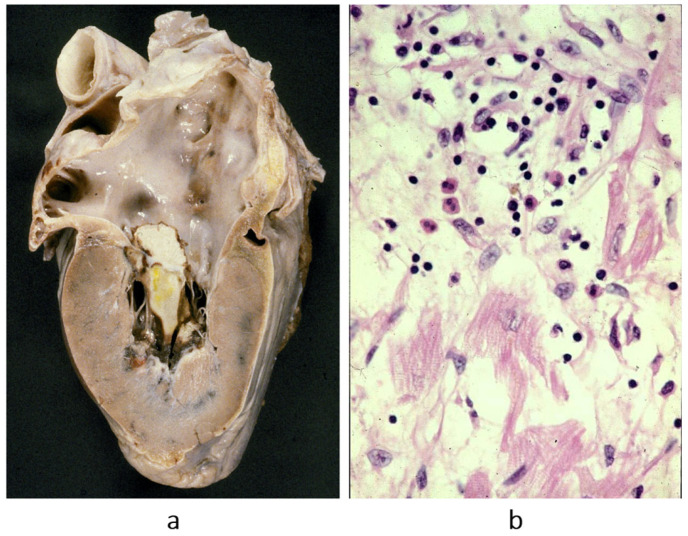 Figure 3
Figure 3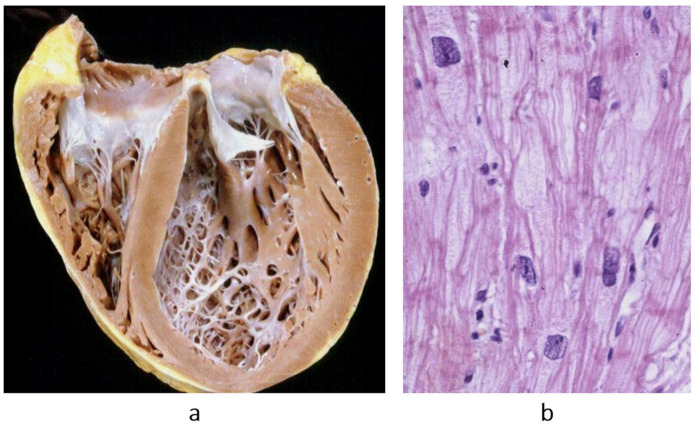 Figure 4
Figure 4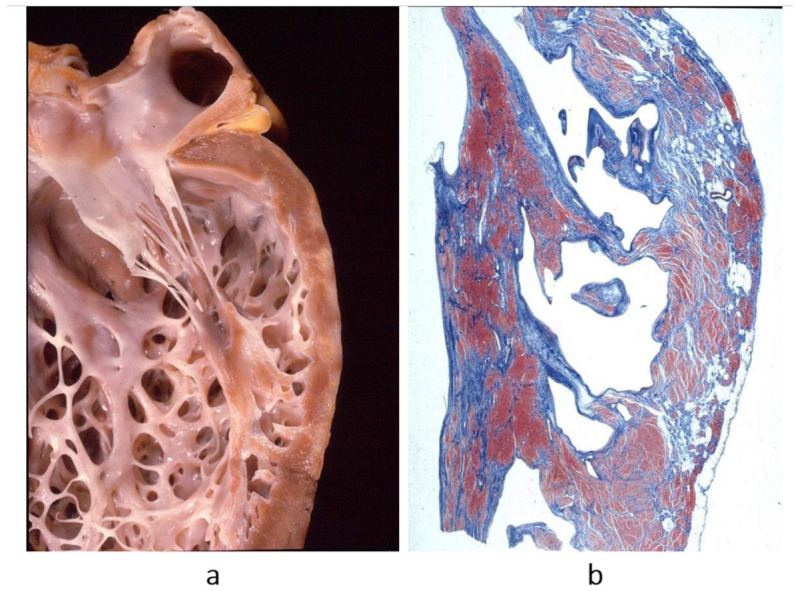 Figure 5
Figure 5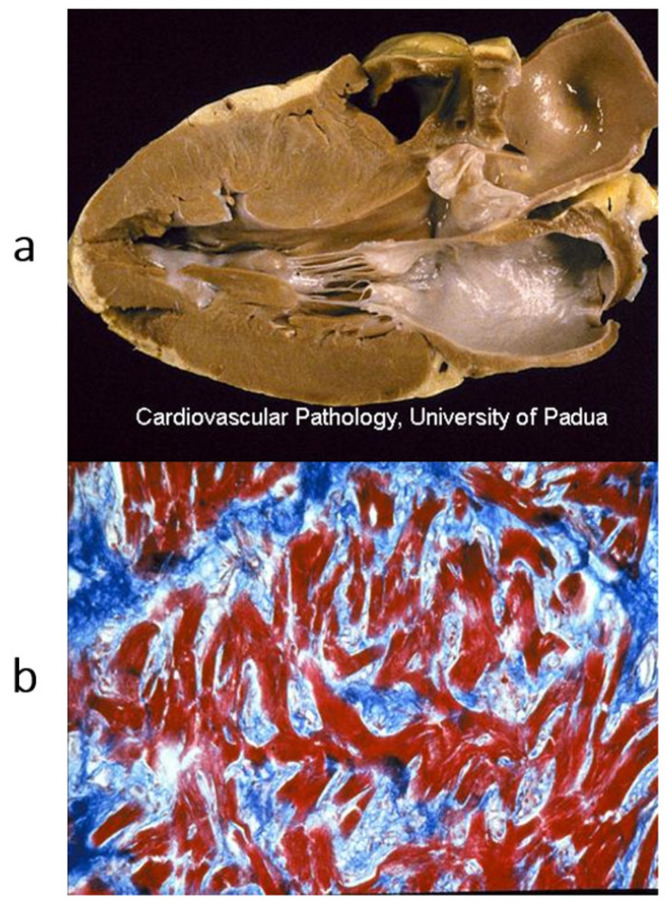 Figure 6
Figure 6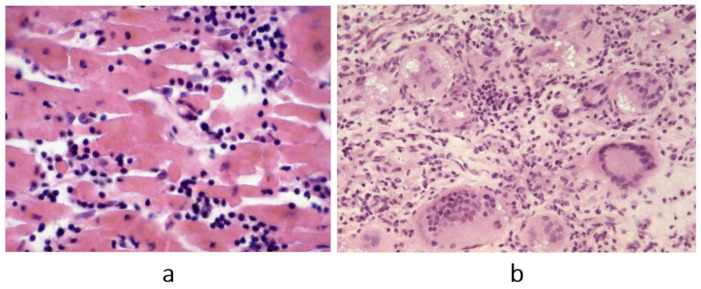 Figure 7
Figure 7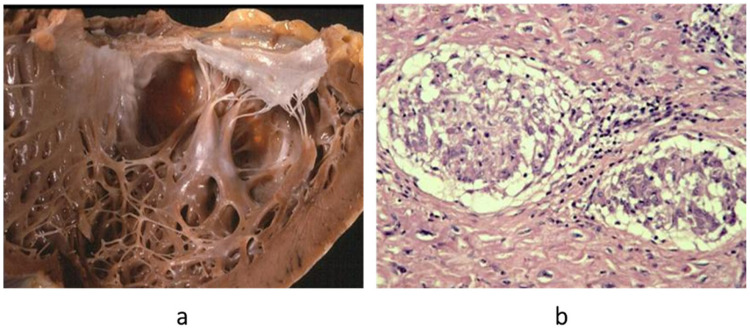 Figure 8
Figure 8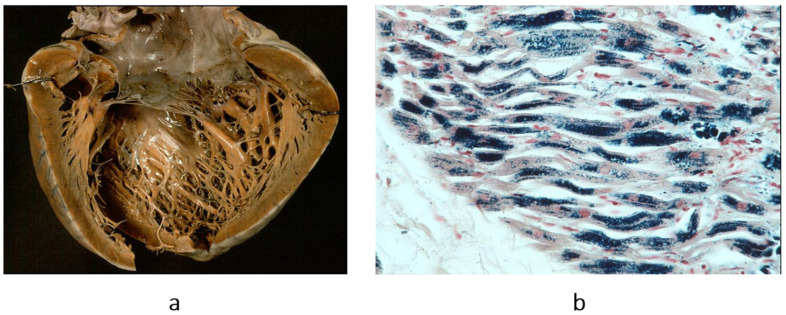 Figure 9
Figure 9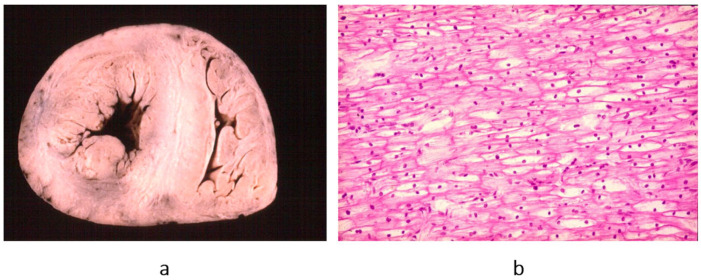 Figure 10
Figure 10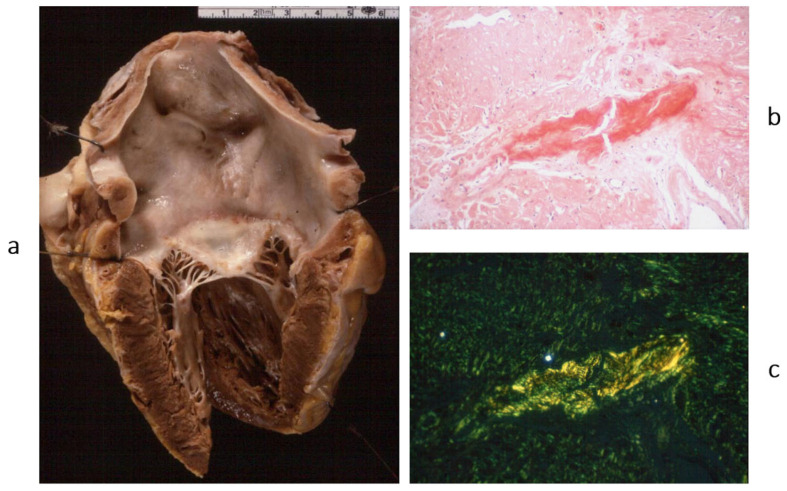 Figure 11
Figure 11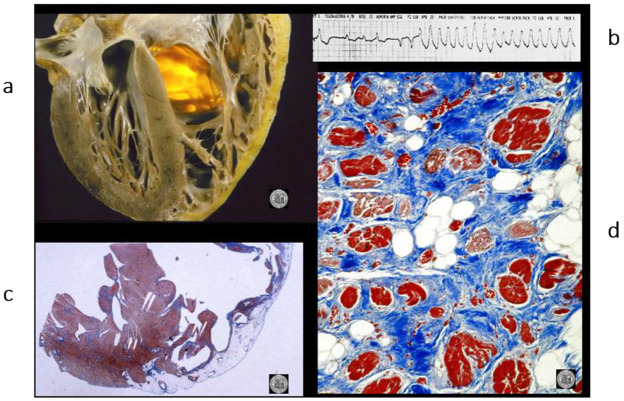 Figure 12
Figure 12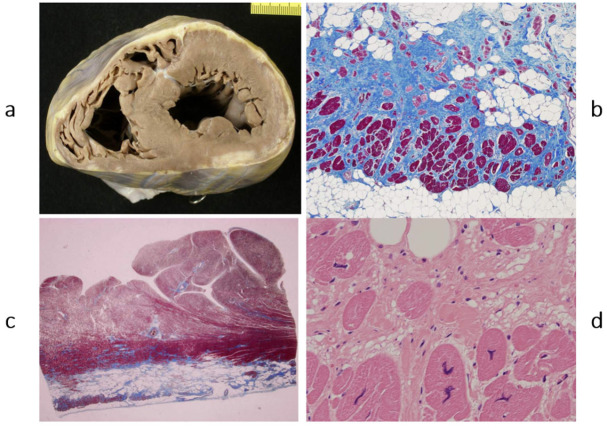 Figure 13
Figure 13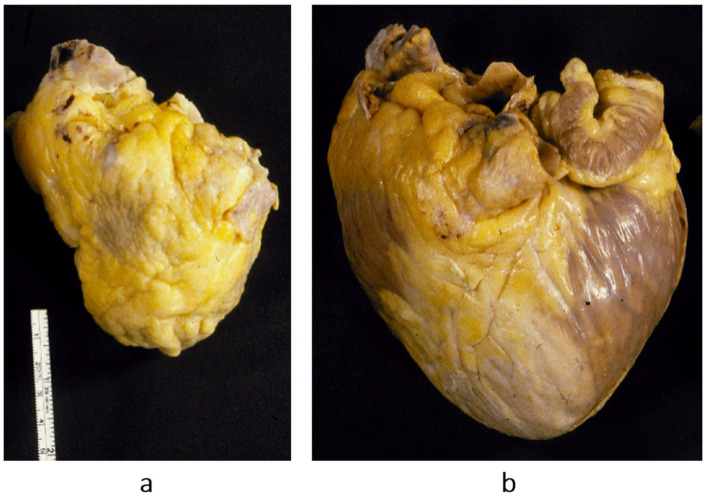 Figure 14
Figure 14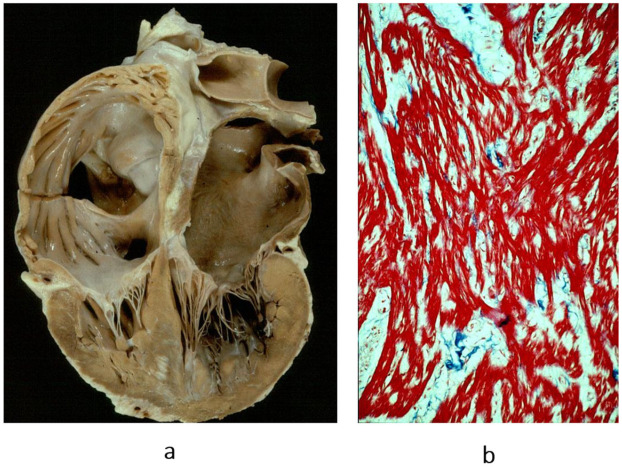 Figure 15
Figure 15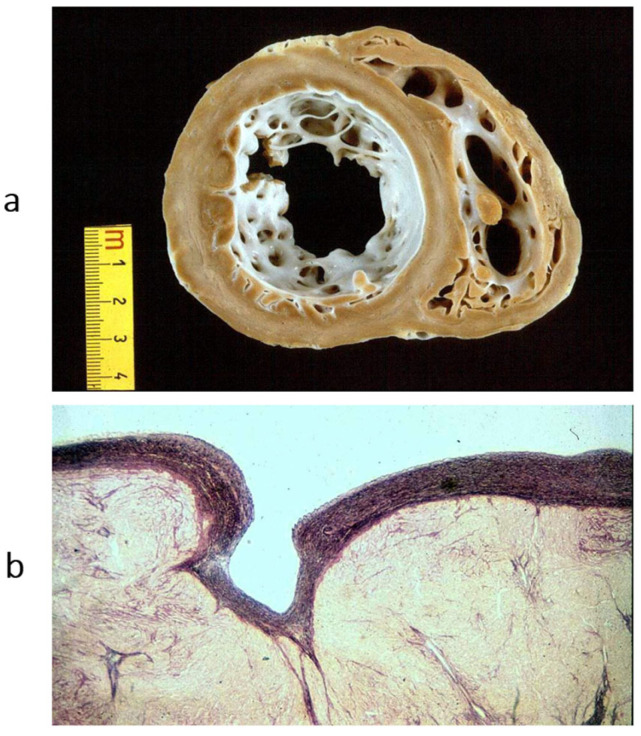 Figure 16
Figure 16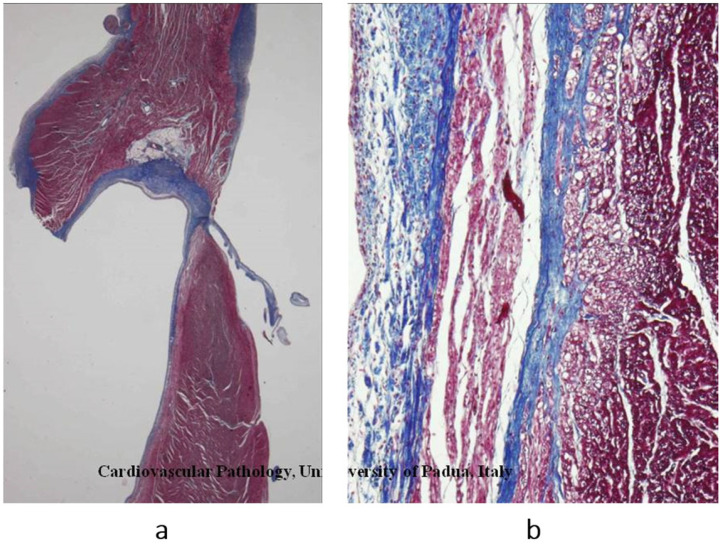 Figure 17
Figure 17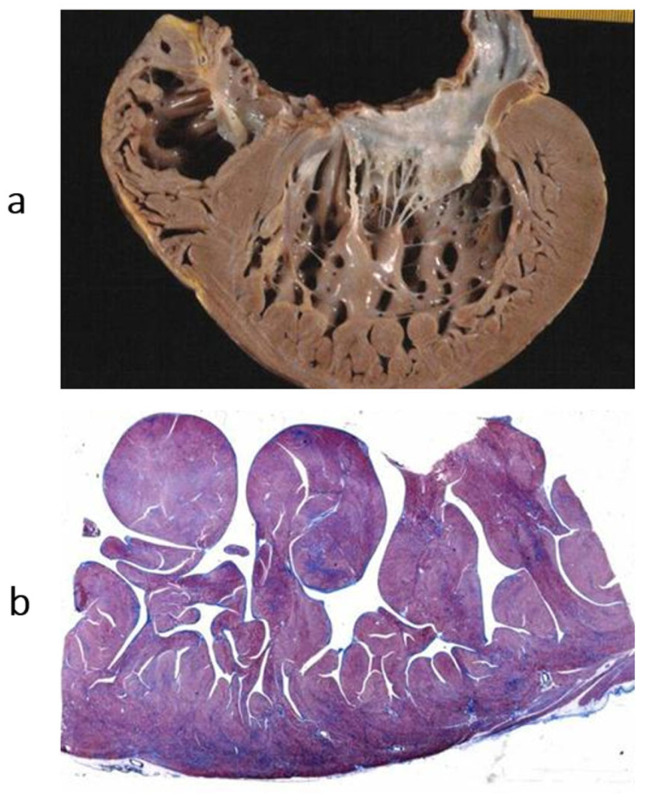 Figure 18
Figure 18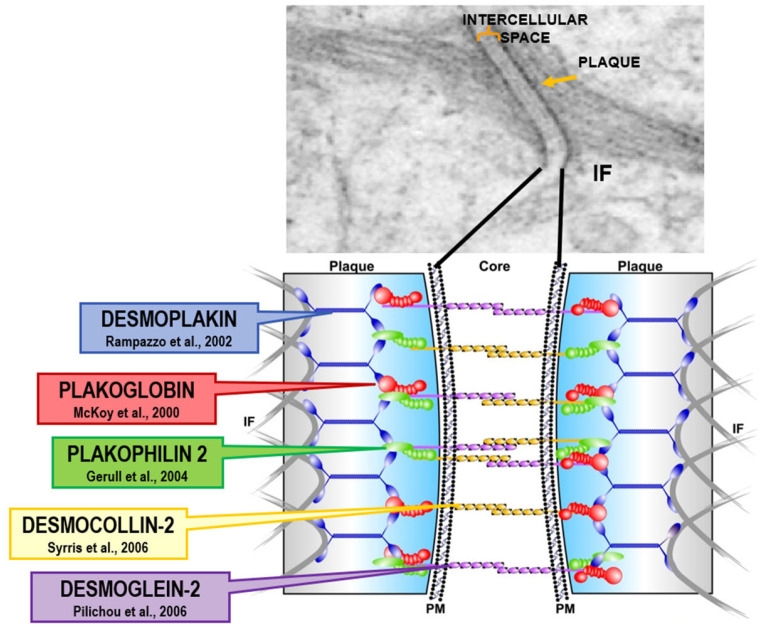 Figure 19
Figure 19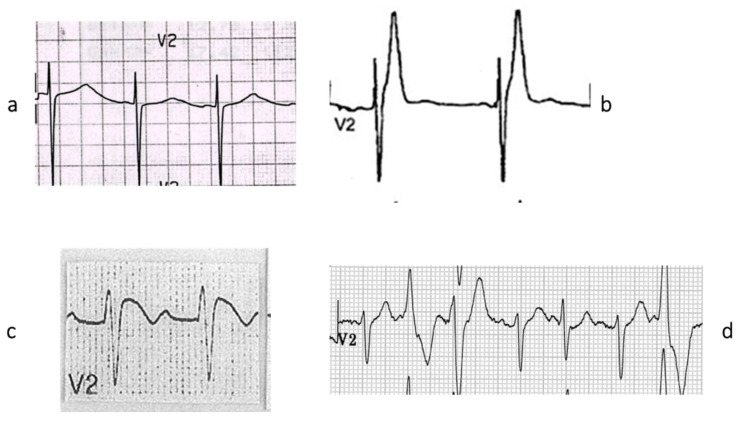 Figure 20
Figure 20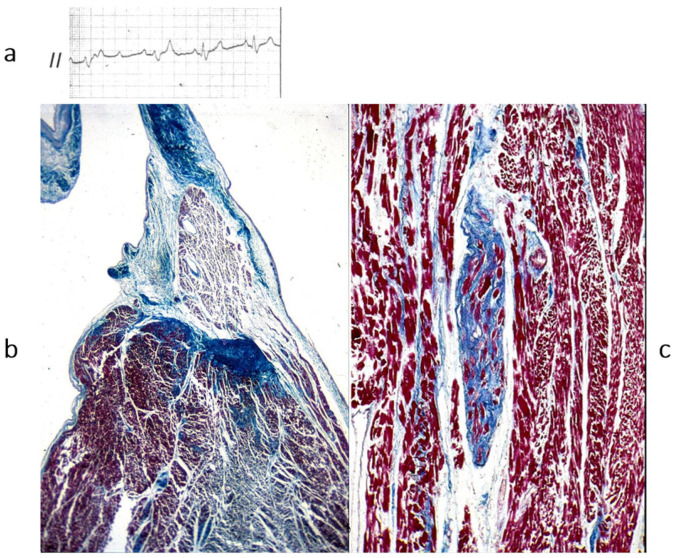 Figure 21
Figure 21 Figure 22
Figure 22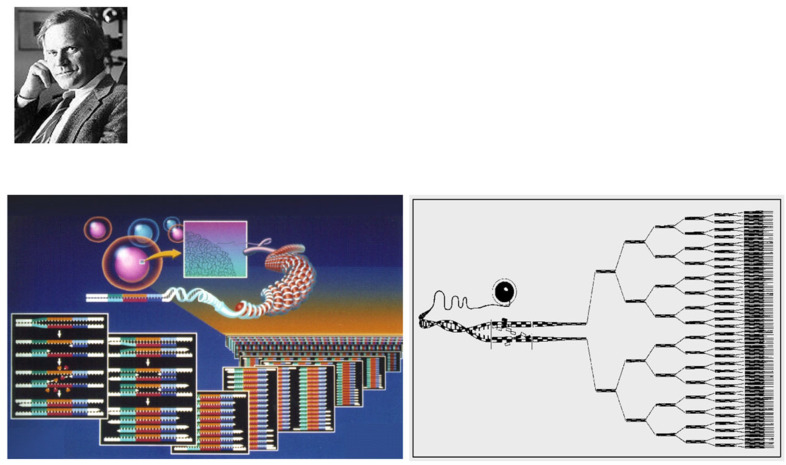 Figure 23
Figure 23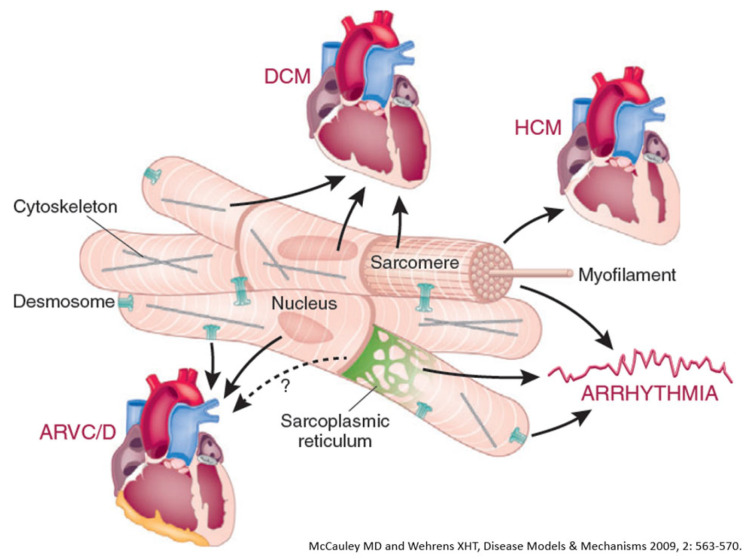 Figure 24
Figure 24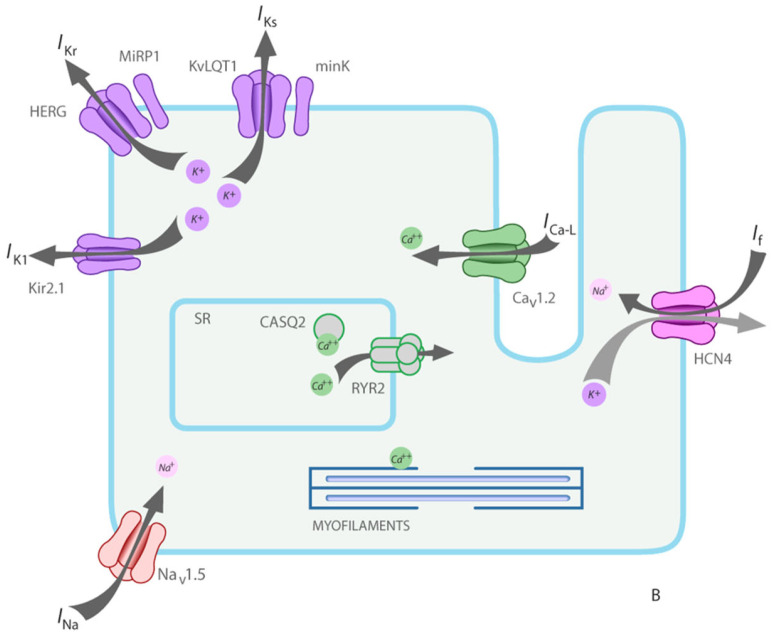 Figure 25
Figure 25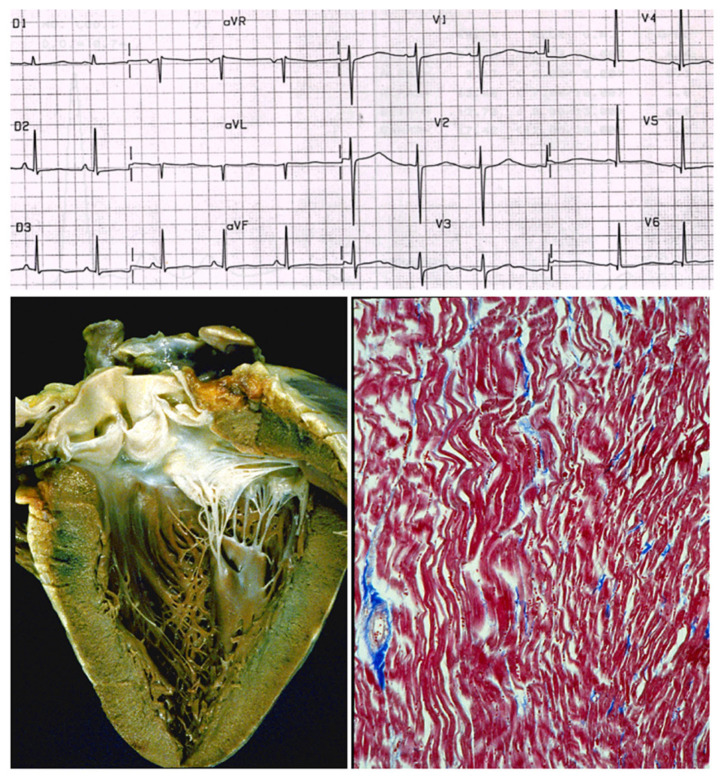 Figure 26
Figure 26- —Cardio-Cerebro-Vascular Pathology Registry, Veneto Region, Venice
- —ARCA Foundation, Padua, Italy
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiomyopathy and Myosin Studies · Cardiovascular Effects of Exercise · Viral Infections and Immunology Research
1. Introduction
In 1980, John F. Goodwin (1918–2001) (Figure 1), Professor of Cardiology at the Westminster Hospital in London and chair of a committee of the WHO, introduced the term cardiomyopathy to describe heart muscle disease of unknown cause and specific heart muscle disease in which the myocardium is associated with multiorgan involvement.
Goodwin’s paper was published in the British Heart Journal with the title “Report of the WHO/ISFC task force on the definition and classification of cardiomyopathies” [2]. Disorders of the myocardium due to systemic or pulmonary hypertension, coronary artery, cardiac valves and congenital heart diseases were ruled out.
2. Historical Overview
Cardiomyopathies were initially classified as dilated (Figure 2a), hypertrophic (Figure 2b) and restrictive–obliterative (Figure 2c), the latter meaning obliteration of ventricular cavities by eosinophilic endomyocardial inflammation with thrombosis (Loeffler heart disease) (Figure 3). Eosinophilia may be due to eosinophil leukemia or due to intestinal infection by helminths, known in Africa by the name of Davies disease.
Concerning dilated cardiomyopathy, ventricular dilatation (Figure 2a and Figure 4a) can be due to poor contractility caused by myocytolysis (Figure 4b) or previous viral myocarditis with scarring from necrosis (Figure 5).
As for hypertrophic cardiomyopathy (Figure 2b), it is characterized by asymmetric hypertrophy of the left ventricle, with myocardial disarray at histology (Figure 6), a bizarre tridimensional arrangement of cardiomyocytes initially interpreted as cardiac hamartoma [1].
3. Pathophysiological Classification of Specific Cardiomyopathies
Among specific heart muscle diseases, a series of morbid entities were included [2]:
- Myocarditis [5].
- Metabolic diseases (endocrine, storage, amyloid).
- General system diseases (connective tissue disorders, sarcoidosis, leukemia).
- Heredofamilial (muscular dystrophies, neuromuscular disorders).
- Sensitivities and toxic reactions (sulphonamides, alcohol, anthracyclines, irradiation).
Regarding inflammatory myocardial diseases, Fiedler myocarditis [6] (Figure 7) was named “isolated”, because only the heart was affected, and “interstitial” because the myocardial interstitium was infiltrated by inflammatory cells, unlike diphtheric myocarditis, in which the damage was only parenchymal. In Fiedler myocarditis, the lymphocytic inflammatory infiltrate (Figure 7a) was most probably due to a viral infection; however, at that time, molecular diagnosis for viral detection was not yet available. A giant cell pattern was also observed in Fiedler myocarditis (Figure 7b), probably ascribable to immune reaction. Sarcoid myocarditis, with non-caseous granuloma, is also most probably immune-related in terms of pathophysiology (Figure 8). It may involve the lymph nodes and other organs like the lungs.
4. Storage- and Interstitial-Specific Cardiomyopathies
Intracellular storage occurs in hemochromatosis (Figure 9), glycogenosis (Figure 10) and Fabry’s disease. Amyloidosis deposits occur (Figure 11) in the interstitium and in the small vessel wall.
5. Chronology of WHO Classification
After the WHO classification of 1980 [7], another two cardiomyopathies were discovered: arrhythmogenic right ventricular [8] (Figure 12) and, more recently, arrhythmogenic left ventricular (Figure 13). They are characterized by fibrofatty replacement of the myocardium in the subepicardium and are at high risk of arrhythmic sudden death.
The second novel type of cardiomyopathy is restrictive, not obliterative, characterized by stiff diastole with poor ventricular relaxation and by atrial dilatation in the absence of ventricular hypertrophy. The heart is small, and the diastolic ventricular filling is hindered by severe congestive heart failure. When compared with dilated cardiomyopathy, restrictive cardiomyopathy represents the paradox of a small heart requiring transplantation (Figure 14). The histology of the ventricular myocardium shows myocardial disarray, like in hypertrophic cardiomyopathy (Figure 15b). Myocardial disarray accounts for impairment of diastolic ventricular filling. This is the reason why both cardiomyopathies are characterized by atrial fibrillation. Restrictive cardiomyopathy is nowadays jokingly called “hypertrophic cardiomyopathy without hypertrophy”.
The discovery of arrhythmogenic cardiomyopathy [8] and restrictive cardiomyopathy [1] required a review of the WHO classification. In June 1995, the WHO committee met in Genève, chaired by Paul Richardson. A new definition and classification was advanced [9].
6. Storytelling of Definition and Classification of Cardiomyopathies
Table 1 and Table 2 compare the definitions and classifications of 1980 vs. 1996. As for the definition of cardiomyopathy, it was changed from “heart muscle disease of unknown cause” to “disease of the myocardium associated with cardiac dysfunction”. And the definition of specific heart disease was changed in specific cardiomyopathy, so that the term cardiomyopathy was employed for any heart muscle disease (Table 1).
New entities (arrhythmogenic and restrictive non obliterative) were added to the cardiomyopathy classification (Table 2). The new classification was published in Circulation in 1996 [9].
Some cardiomyopathies remained unclassified (Table 3).
Endocardial fibroelastosis (=dilated cardiomyopathy in children) (Figure 16) appears among unclassified cardiomyopathies both in 1980 and 1995 (Table 3).
More recently, in 1997, the aetiology of endocardial fibroelastosis was determined to be an infection in the uterus due to mumps virus of the myocardium thanks to molecular investigation [10,11].
Histiocytoid cardiomyopathy was definitively interpreted as a tumour of Purkinje cells (Purkinjoma) (Figure 17) [12].
In 1996, a non-compacted left ventricular myocardium (Figure 18) was still considered an unclassified cardiomyopathy. Lack of ventricular wall compaction is an embryological defect, so the disease should be considered a congenital heart disease [13].
Nowadays, Fiedler’s myocarditis [6] is considered a cardiomyopathy, as any other viral or immunological myocarditis.
In 2006, the definition and classification of cardiomyopathies also attracted the interest of the American Heart Association [15]. Table 4 reports the 2006 AHA definition of cardiomyopathies. The goals of the 2006 AHA classification were as follows:
- Electrical heart dysfunction is a myocardial dysfunction, so channelopathies are cardiomyopathies.
- When myocardial dysfunction is the consequence of other cardiovascular diseases (valve, hypertension, congenital, coronary artery), it is excluded from the classification of cardiomyopathies.
- Myocarditis is a cardiomyopathy.
Cardiomyopathies are a major cause of severe heart failure requiring cardiac transplantation (Table 5). In the experience (1985–2015) of the University of Padua, cardiomyopathies accounted for 51.4% of heart recipients (Table 5) [3].
Among cases of sudden death in 650 young people, 31.3% of patients died because they were affected by cardiomyopathies: hypertrophic cardiomyopathy, arrhythmogenic cardiomyopathy and myocarditis (Table 6) [3].
7. Genetic Background of Cardiomyopathies
Cardiomyopathies are usually genetically determined. Hippocrates stated that diseases may be handed down from parents to offspring from the very moment of conception. Table 7 reports the classification of inherited cardiomyopathies caused by defective genes and wrong-coded proteins: cytoskeleton, sarcomere, desmosome, and ion channels.
Dilated cardiomyopathy is mostly related to gene defects of cytoskeleton proteins of the nuclear and cell membranes. Missense mutation of lamin A/C and truncation of titin give origin to genetically determined cardiomyopathies, the former also accounting for AV conduction disturbances.Hypertrophic and restrictive cardiomyopathy are both due to mutations of genes coding sarcomere proteins [16,17].Arrhythmogenic cardiomyopathy is the consequence of mutations of genes coding desmosome proteins (Figure 19) [18].Long QT (Figure 20a), short QT (Figure 20b), Brugada syndrome with non-ischemic ST elevation (Figure 20c), polymorphic catecholaminergic ventricular tachycardia (Figure 20d) and Lenegre disease (Figure 21) are ion channel and ryanodine receptor cardiomyopathies.
Also, complete AV block may be inherited, with the name of Lenegre disease (Figure 21).
Thus, mutations of sodium “channel I5” account for long QT, Brugada syndrome and Lenegre disease. The latter is an example of a genetically determined cardiomyopathy of the conduction tissue (Figure 21).
They should be considered cardiomyopathies, regardless of the type of cardiac dysfunction: contractile, electric, intercellular junction, electromechanical association or electric stimulus conduction.
Refs. [17,18,20,21,22,23,24,25,26,27,28] deal with the discoveries of the genetic background of cardiomyopathies. A proposal was advanced of a genetic classification for inherited cardiomyopathies.
8. Is It Time for Gene Therapy?
Genetically determined cardiomyopathies are caused by mutations or deletions of DNA.
Recent studies suggested that gene therapy may be a potent molecular option for the treatment of genetically determined cardiomyopathies, like for hypertrophic cardiomyopathy due to myosin mutations and for catecholaminergic polymorphic ventricular tachycardia. Gene therapy is based upon the introduction of genetic material into cardiomyocytes.
There are several strategies involving DNA and RNA messengers.
9. DNA Action Strategies for Genetic Therapy [29]
Gene replacement by viral vector, with the delivery of a healthy gene copy, introduced into the cardiomyocyte by a viral vector. DNA adenovirus is the most used. However, adenovirus is a frequent etiology of viral myocarditis, so using this vector may result in a huge inflammatory response, with a risk of iatrogenic death.Modification of signal pathways.Inactivation of mutant gene by cleavage of the endogenous DNA to prevent its expression.Mutation repair by restoring healthy genes after cleavage.Oligonucleotides are short DNA or RNA molecules that can be used to perturb gene expression in target cells. They may be expressed by viral vectors or chemically synthesized and delivered systemically.Modified RNA messengers may be synthesized by in vitro transcription using modified nucleotides, designed to allow them to enter inside the cardiomyocytes without immune reaction and rapid translation into protein.
10. Final Reflections
Thanks to the discovery of DNA by James Watson and Francis Crick (Figure 22) [30] and the invention of polymerase chain reaction by Kary Banks Mullis (Figure 23) [31], molecular diagnosis of inherited heart disease is feasible even at autopsy and can be specific. Gene therapy is a life-saving novelty that can prevent the transmission of inherited disease to offspring.
Just like vaccinations led to the disappearance of lethal infective diseases like smallpox, with gene therapy, diseases transmissible to the offspring may also disappear.
Indeed, the dream is the disappearance of every genetic disease in the future, including genetically determined cardiomyopathies (Figure 24), both with and without (Figure 25 and Figure 26) structural defects, both of which pose an equal risk of sudden death.
In 1894, Karl von Rokitansky and Rudolph Virchow attended a meeting on “Morgagni and the Anatomic Concept” in Rome. Virchow raised the following questions: “…Any anatomic modification is material, but is any material modification anatomic? Why not molecular? Can a profound molecular modification occur in the setting of an apparently normal structure? These modifications belong more to physiology than to anatomy, they are functional-dynamic… the method of investigation will never be morphological”.
Refs. [34,35,36,37,38,39] provide further contributions on gene therapy for inherited and genetically determined cardiomyopathies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Thiene G. Calore C. De Gaspari M. Basso C. Storytelling of Hypertrophic Cardiomyopathy Discovery J. Cardiovasc. Dev. Dis.20241130010.3390/jcdd 1110030039452271 PMC 11508572 · doi ↗ · pubmed ↗
- 2Goodwin J.F. The frontiers of cardiomyopathy Br. Heart J.19824811810.1136/hrt.48.1.16123334 PMC 481195 · doi ↗ · pubmed ↗
- 3Mc Kenna W.J. Maron B.J. Thiene G. Classification, Epidemiology, and Global Burden of Cardiomyopathies Circ. Res.201712172273010.1161/CIRCRESAHA.117.30971128912179 · doi ↗ · pubmed ↗
- 4Thiene G. Corrado D. Basso C. Sudden Cardiac Death in the Young and Athletes Springer Milano, Italy 2016978-88-470-5775-3
- 5Thiene G. Storytelling of Myocarditis Biomedicines 20241283210.3390/biomedicines 1204083238672187 PMC 11048135 · doi ↗ · pubmed ↗
- 6Fiedler KLA Über akute interstitielle Myokarditis. Dresden, W. Baensch Zentralblatt für innere Medizin 190021212213
- 7Report of the WHO/ISFC task force on the definition classification of cardiomyopathies Br. Heart J.19804467267310.1136/hrt.44.6.6727459150 PMC 482464 · doi ↗ · pubmed ↗
- 8Thiene G. Nava A. Corrado D. Rossi L. Pennelli N. Right ventricular cardiomyopathy and sudden death in young people N. Engl. J. Med.198831812913310.1056/NEJM 1988012131803013336399 · doi ↗ · pubmed ↗