Decoding the Impacts of Mating Behavior on Ovarian Development in Mud Crab (Scylla paramamosain, Estampador 1949): Insights from SMRT RNA-seq
Chenyang Wu, Sadek Md Abu, Xiyi Zhou, Yang Yu, Mhd Ikhwanuddin, Waqas Waqas, Hongyu Ma

TL;DR
This study explores how mating affects ovarian development in mud crabs by analyzing gene expression and splicing changes, revealing key molecular pathways involved in reproduction.
Contribution
The study provides the first comprehensive view of transcriptional and splicing dynamics in post-mating ovarian maturation in mud crabs.
Findings
Mating triggers major transcriptional reprogramming in the ovary, with serotonin and dopamine signaling acting as opposing regulators of oocyte maturation.
Later stages of ovarian development involve energy metabolism, lipid mobilization, and extracellular matrix remodeling to support yolk accumulation.
Enhanced antioxidant defenses are observed, indicating the importance of maintaining redox balance during rapid ovarian development.
Abstract
In crustaceans, pubertal molting marks the shift from growth to reproduction, and mating promotes ovarian maturation. However, the molecular events driving this transition remain unclear. In this study, we used full-length transcriptome sequencing to investigate gene expression and alternative splicing changes in the mud crab (Scylla paramamosain) after mating. Our results showed that mating triggered major transcriptional reprogramming in the ovary. Early responses involved serotonin and dopamine signaling, which act as opposing regulators of oocyte maturation. Later stages were characterized by activation of energy metabolism, lipid mobilization, and extracellular matrix remodeling to support yolk accumulation and oocyte growth. We also observed enhanced antioxidant defense, indicating the importance of maintaining redox balance during rapid ovarian development. These findings provide…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
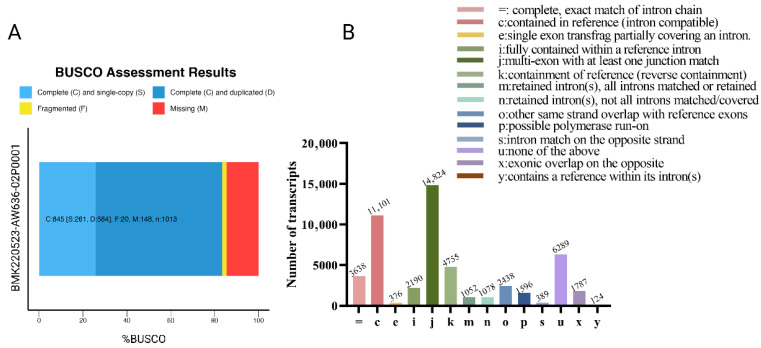 Figure 1
Figure 1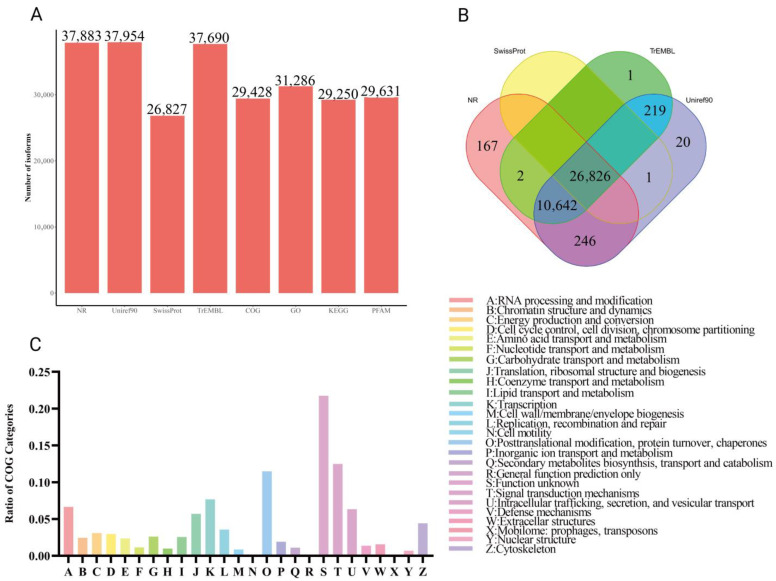 Figure 2
Figure 2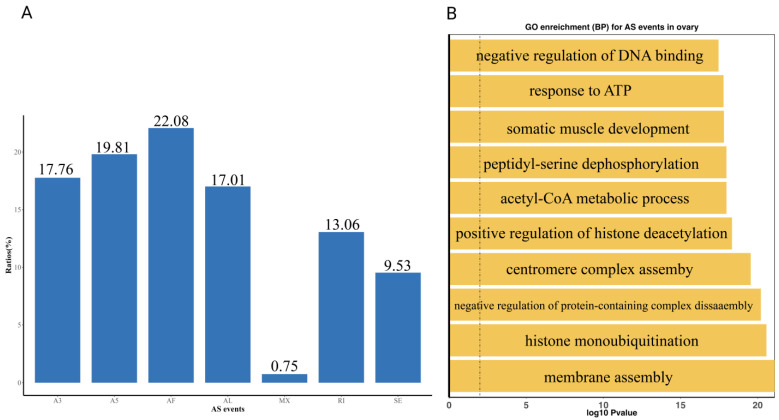 Figure 3
Figure 3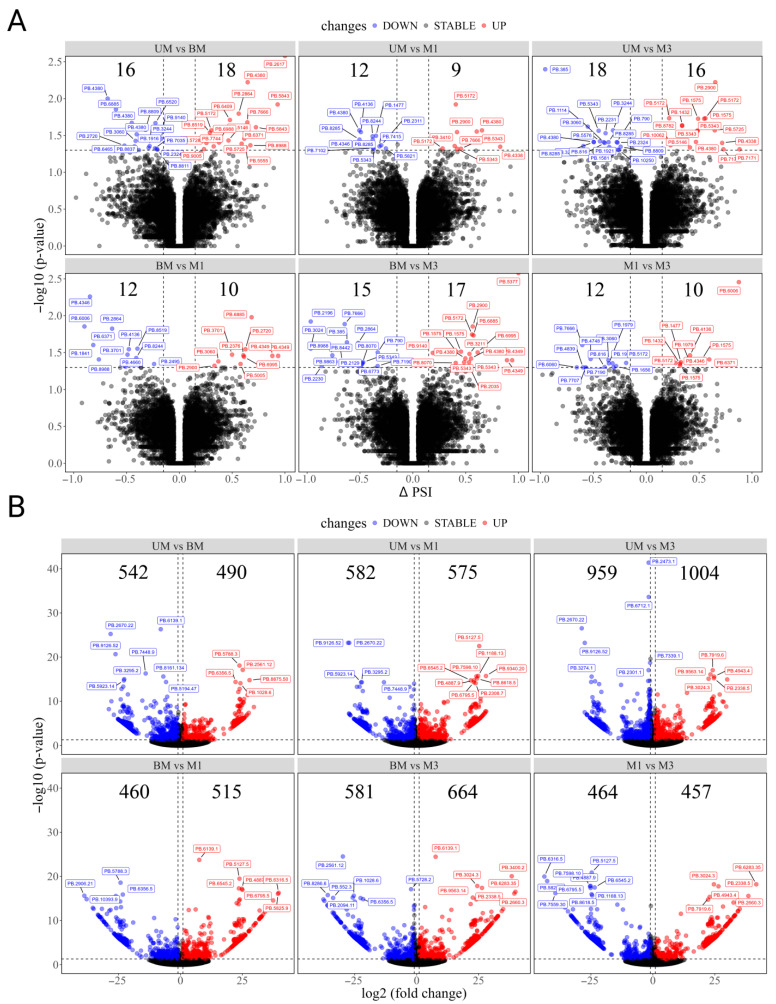 Figure 4
Figure 4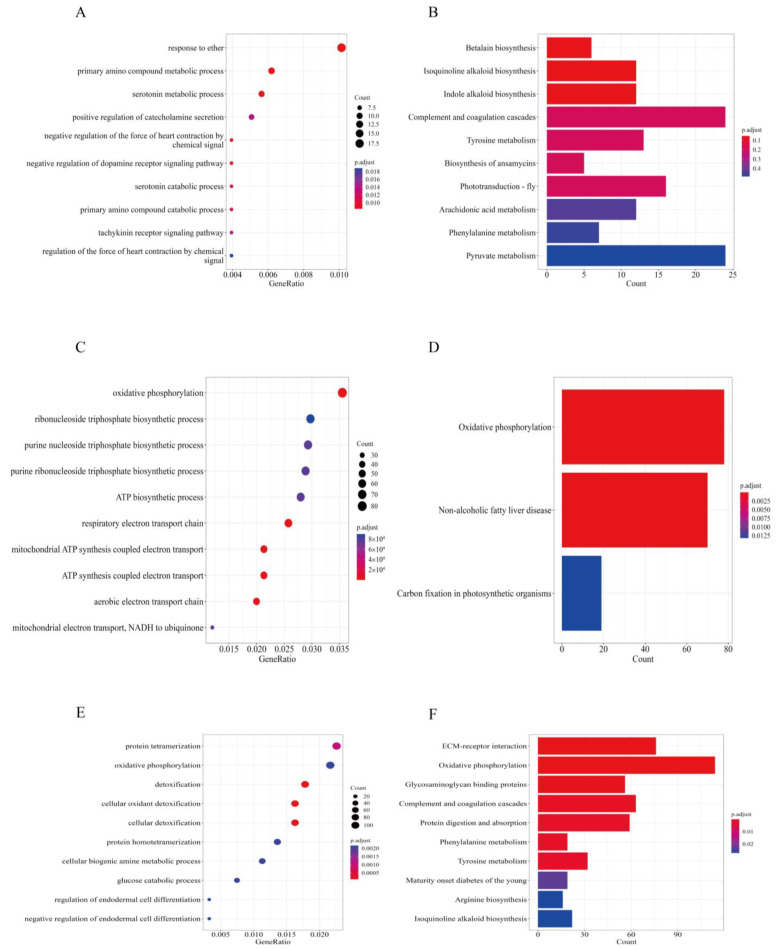 Figure 5
Figure 5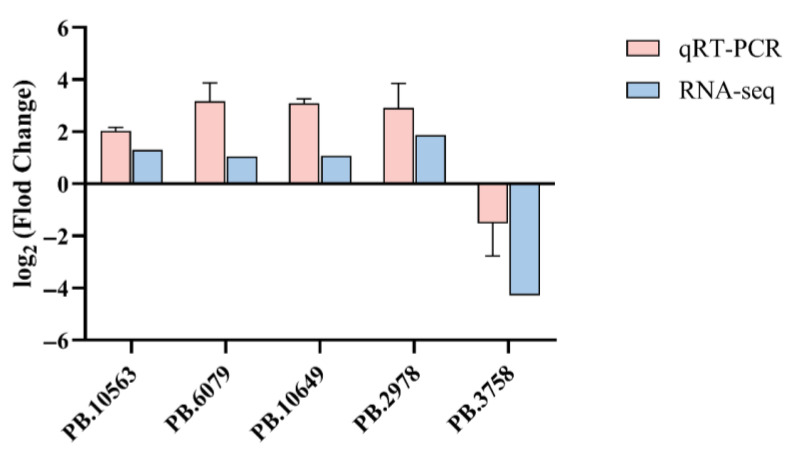 Figure 6
Figure 6- —National Natural Science Foundation of China
- —Agricultural and Rural Department of Guangdong Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCrustacean biology and ecology · Aquaculture Nutrition and Growth · Invertebrate Immune Response Mechanisms
1. Introduction
The genus Scylla includes commercially important aquaculture crab species cultured across Indo-Pacific regions [1,2]. Among the Scylla crabs, the mud crab (Scylla paramamosain, Estampador 1949), a key species, has emerged as one of the most economically important crustaceans and is widely cultured across China and Vietnam [3,4]. S. paramamosain is valued for its rapid growth, high fecundity, and strong market demand. In the year 2023, its production was recorded as 157,012 tons in China, showcasing its significance in crustacean aquaculture [5]. Female crabs are important from both ecological and aquaculture perspectives due to their economic value and reproduction capabilities [6]. In hatcheries, female crabs are the primary source of larval production, while their developed ovaries make them highly sought in seafood markets [7,8]. Additionally, their reproductive traits serve as sensitive indicators of environmental stress, making female crabs key models in environmental and molecular research [9]. Ovarian development is a complex process regulated by the neuroendocrine network involving various hormones and enzymes such as ecdysone and vitellogenin [10]. Ovarian development in female crabs after mating demands a higher energy and material supply than unmated individuals [11]. This process involves a multifaceted interplay of molecular entities, including genes, hormones, enzymes, lipids, and proteins, forming a complicated interactive network [12]. However, the molecular profiling of post-mated female mud crabs is largely unexplored, leaving a critical gap in the literature on how mating behavior influences the molecular dynamics of ovarian development.
In mud crabs, ovarian development follows a gradual process consisting of five stages: starting from the undeveloped phase (Stage I), progressing through pre-vitellogenic (Stage II), early and late vitellogenic stages (Stages III and IV), and culminating in the fully mature stage (Stage V) [13]. Before undergoing pubertal molt and mating, mud crabs usually have ovaries at Stage II, which are still small and appear translucent to milky white [14]. In the early stages of ovarian development, oogonia actively proliferate and then begin to differentiate into primary oocytes [15]. As Stage II progresses, follicular cells begin to proliferate around the cytoplasm of the developing oocyte [16]. Following mating, the mud crab undergoes rapid ovarian development and yolk accumulation [17]. Thus, mating behavior serves as a potential catalyst of ovary development and vitellogenesis in female crabs following pubertal molt [18]. Eubrachyuran crabs exhibit six mating strategies, differentiated by the timing of mating relative to molting (soft- or hard-shell), growth pattern (determinate or indeterminate), and seminal receptacle position (dorsal or ventral) [19]. The mud crabs mate during their soft-shell stage and has dorsal seminal receptacles. In the genus Scylla, mating involves courtship, extended guarding, sperm plugs, and the absence of hinged vulval coverings [20].
Next-generation sequencing (NGS) has made it possible to quickly and accurately analyze genetic material, opening new doors for understanding how genes function and interact [21,22]. NGS has enabled fast, affordable DNA sequencing, driving the development of tools like RNA-seq, exome sequencing, ChIP-seq, miRNA-seq, RAD-seq, and small RNA sequencing to tackle diverse biological [23,24,25]. However, short-read NGS poses limitations for accurate transcript reconstruction in eukaryotes due to complex splicing and transcript isoforms [26]. This complexity stems from the ability of a single gene to produce multiple isoforms through alternative transcription start sites and RNA processing. Long-read sequencing overcomes this limitation by capturing full-length isoforms, enabling more accurate transcriptome analysis [27]. SMRT RNA-seq produces highly accurate long reads, enabling analysis of alternative splicing, polyadenylation, genome annotation, fusion transcripts, isoform phasing, and long noncoding RNAs [28]. Its real-time, PCR-free workflow minimizes bias, reduces costs, and lowers computational requirements [29]. Full-length transcripts refine genome annotation by precisely mapping gene structures, regulatory elements, and coding regions, facilitating detailed isoform analysis across the transcriptome [30].
In Portunus trituberculatus, the GSI of mated females was significantly higher than that of unmated females. Histological observations revealed the presence of exogenous vitellogenic oocytes in mated females, whereas the predominant oocytes in unmated females were previtellogenic oocytes and endogenous vitellogenic oocytes [31]. Previously, Yu et al. [32] reported that ovarian development in unmated mud crabs is prolonged, taking up to 60 days with smaller oocytes, while mated females reach stage III (proliferative stage) within 23 days and develop larger oocytes, indicating that mating behavior triggers ovarian maturation. However, the molecular mechanism linking mating behavior to ovarian development remains unclear. In this study, we employed full-length transcriptome analysis using SMRT RNA-seq to explore differentially expressed genes, alternative splicing events, and regulatory pathways involved in ovarian development in Scylla paramamosain before and post-mating stages. These findings provide novel insights into the molecular mechanisms by which mating behavior regulates ovarian maturation in Scylla paramamosain. These insights are expected to advance fundamental research on reproductive regulation and to inform the development of improved broodstock management strategies in aquaculture.
2. Materials and Methods
2.1. Crabs Collection and Mating
A total of 80 female and 30 male mud crabs were collected from a local seafood market in Chaozhou city (23.26° N, 116.54° E) of Guangdong Province in China, with detail information in our previous research [31]. Briefly, crabs were reared individually in water tanks containing filtered seawater maintained at 29 °C and fed twice daily with clams (Sinonovacula constricta). Pubertal molting and courtship were monitored using a real-time video surveillance system [33]. Artificial mating between male and female crabs was carried out, following the method of Fazhan et al. [17]. Immediately after reproductive molt, female crabs were paired with potential males in dark and oxygenated mating boxes for 1–2 days to facilitate copulation. Clean water and food were provided to maintain water quality and reduce the risk of male cannibalism during mating. To ensure consistency in the artificial mating process, all pairings were conducted under identical environmental conditions. The procedures were standardized and carried out by the same operator to minimize variation among groups. Each female crab was used only once for mating, while the male crabs were used repeatedly. Additionally, four experimental groups were established to represent key stages of reproductive development: (1) BM—before pubertal molting, (2) UM—unmated females exhibiting courtship behavior without successful copulation, (3) M1—females at 1-day post-mating, and (4) M3—females at 3 days post-mating. Mating was confirmed by detecting spermatophores in the seminal receptacles of female crabs anesthetized on ice [34,35]. The crabs were then dissected, and approximately 50 mg of ovarian tissue was collected and preserved in RNA Keeper Tissue Stabilizer (Vazyme Biotech Co., Ltd., Nanjing, China). Following the incubation at 4 °C for 24 h, the tissue samples were stored −80 °C until RNA sequencing. A total of 12 ovarian tissue samples—three biological replicates each from the BM, UM, M1, and M3 groups—were subjected to RNA sequencing.
2.2. RNA Extraction and Sequencing
Total RNA was extracted using the TRIzol Reagent kit (ComWin Biotech, Guangzhou, China). RNA quality was assessed by measuring concentration and integrity using an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA), and a Nanodrop 2000 (Agilent Technologies, CA, USA). Only high-quality RNA samples with a RNA integrity number (RIN) ≥ 8 and a 28S/18S ratio ≥ 1.5 were selected for subsequent library construction. For SMRT sequencing, libraries were constructed and sequenced using the PacBio Sequel platform (v Sequel II). Briefly, PacBio single-molecule long reads were processed using RS_IsoSeq (v2.3) in the SMRT Analysis package (v2.3.0.140936.p4.150482) via command line to obtain insert reads. Full-length transcript characterization was performed using the pbtranscript.py script from the same package. The SMRT library was constructed from pooled RNA extracted from all 12 crab ovarian tissue samples. PacBio single-molecule long reads were processed using RS_IsoSeq (v2.3) in the SMRT Analysis package (v2.3.0.140936.p4.150482) via command line to obtain insert reads. Full-length transcript characterization was performed using the pbtranscript.py script from the same package. The Clontech kit was used to identify the 5′ and 3′ primers, with the poly(A) tail upstream of the 3′ primer serving as a key signal for identifying strand-specific full-length reads. Sequencing errors in consensus reads were corrected using LSC 2.0 (https://github.com/Augroup/LSC, accessed on 17 August 2025) with Illumina short reads derived from ovarian tissues of S. paramamosain (parameters: runLSC.py -long_reads SQ_SMRT.fa -short_reads SQ_Illumina.fa -output output). The completeness of the SMRT-based transcriptome assembly was then evaluated using DETONATE and Ex90N50 metrics. For Illumina sequencing, three libraries were prepared per group (BM, UM, M1, and M3) and sequenced on the Illumina HiSeq 2500 platform (v 6000) to generate 150 bp paired-end reads. Library construction and sequencing were carried out by Beijing Biomarker Technologies Co., Ltd., Beijing, China.
2.3. SMRT Sequencing Data Processing
To identify transcript isoforms, we processed the subread sequence data using a structured bioinformatics pipeline. Firstly, circular consensus sequences (CCS) were generated from the raw subreads using ccs (v4.2.0) with parameters set to -minLength 50, -maxLength 15,000, and -minPasses 1 to ensure high-quality reads. Next, full-length (FL) reads were obtained by removing primers and demultiplexing with lima (v1.11.0), applying the -dump-clips and -peek-guess parameters for accurate read processing. To eliminate noise, FL reads were refined using the isoseq3 (v3.3.0) refine module, requiring polyadenylation (-require-polya) and a minimum poly(A) tail length of 20 nucleotides (-min-polya-length 20). These filtered reads were then clustered into unpolished transcript consensus sequences using the same IsoSeq3 refine module with default settings. Finally, the unpolished transcripts were polished using the isoseq3 polish module (default parameters) to generate high- and low-confidence transcript isoforms, ensuring robust isoform identification for downstream analysis.
2.4. Collapsing Redundant Transcripts Isoforms
To reduce redundancy among transcript isoforms, we implemented a two-step collapsing approach. Firstly, high-quality isoforms were aligned to the Scylla paramamosain reference genome [36] using minimap2 (v2.18) (default parameters), generating SAM files [37]. Redundant mapped isoforms were then collapsed using cDNA_Cupcake (https://github.com/Magdoll/cDNA_Cupcake) with parameters (-min_aln_coverage 0.95, -min_aln_identity 0.85, -dun-merge-5-shorter). For unmapped transcripts, we employed Cogent (v8.0) (https://github.com/Magdoll/Cogent) and cDNA_Cupcake (default parameters) to identify distinct gene families. A “fake genome” was constructed by concatenating Cogent-derived contigs, allowing unmapped transcripts to be realigned and collapsed following the same criteria. Finally, CD-HIT (v4.8.1) [38] was applied to both mapped and unmapped isoforms to remove highly similar sequences (identity threshold: 95%), ensuring a non-redundant transcriptome for downstream analysis.
2.5. Completeness and Characteristics Analysis of Reconstructed Transcriptomes
To evaluate the quality and completeness of the full-length transcriptomes, we conducted benchmarking universal single-copy orthologs (BUSCO) (v5.1.3) [39] analysis in transcriptome mode using the Arthropoda lineage dataset (arthropoda_odb10) to assess the representation of evolutionarily conserved single-copy orthologs [40]. The full-length transcripts were then systematically classified by comparing them to the reference genome annotation using gffcompare (v0.12.2) (http://ccb.jhu.edu/software/stringtie/gffcompare.shtml) [41] (Table 1). To identify shared and unique transcripts across the datasets, we performed pairwise comparisons using BLASTn (v2.11.0^+^) with stringent parameters (−e value ≤ 1 × 10^−10^ and percent identity ≥ 95%), where each transcriptome was alternately used as both the reference database and query sequence. This comprehensive analytical approach enabled us to thoroughly characterize the transcriptomes and identify both conserved and sample-specific transcripts.
2.6. Gene Functional Annotation
To elucidate the biological functions of the full-length transcripts, we performed comprehensive gene functional annotation through a multi-step bioinformatics. Firstly, we employed TransDecoder to identify potential open reading frames (ORFs) from the transcripts, selecting the first ORF when multiple candidates were present in a single transcript. These ORFs were then functionally annotated using eggNOG-mapper (v2.1.4) [42] to obtain classifications from multiple databases, including Clusters of Orthologous Groups (COG), Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Protein families database (Pfam). To further validate and expand the functional annotations, we conducted homology searches against four major protein databases (Swiss-Prot, TrEMBL, UniRef90, and NR) using DIAMOND (v2.0.4.142) with stringent parameters (blastp, -outfmt 6, -max_target_seqs 1, -e value 1 × 10^−5^). Finally, all annotation results were consolidated into a comprehensive, tab-delimited summary file to facilitate further functional analyses.
2.7. Alternative Splicing (AS) Events Analysis
Alternative splicing (AS) events were analyzed using SUPPA2 software (v 2.4) [43], and seven major types of AS events were identified using the generateEvents function of SUPPA2 with default parameters. These included skipped exon (SE), mutually exclusive exon (MX), alternative 5′ splice site (A5), alternative 3′ splice site (A3), retained intron (RI), alternative first exon (AF), and alternative last exon (AL). The distribution and frequency of these AS events were compared across different mating groups to identify potential splicing differences linked to genetic background. Furthermore, Gene Ontology (GO) enrichment analysis of genes associated with AS events was conducted using the clusterProfiler package (v 4.0.4), based on functional annotations obtained through eggNOG-mapper.
2.8. Quantification of Identified Transcripts
To investigate differential alternative splicing (AS) events among the BM, UM, M1, and M3 groups, subread sequences from each group were processed to reconstruct group-specific transcriptomes, following the pipeline previously described. Transcript and gene expression quantification were then performed using Salmon (mapping-based mode) [44]. Firstly, a reference transcriptome index was generated by constructing a k-mer hash (k = 31) from the reconstructed transcript sequences. Subsequently, clean RNA-seq reads were aligned to the corresponding group-specific transcriptomes using Salmon’s quant module with the parameters -l IU and -validateMappings for fast and accurate alignment. Finally, transcript abundance was estimated using the quantmerge module, which produced transcript per million (TPM) values for each sample. These TPM values were subsequently used to calculate percent spliced-in (PSI) values, providing a quantitative measure of alternative splicing across the different groups.
2.9. Differential Alternative Splicing (DAS) Events, Differential Expressed Transcripts (DETs) and Their Enrichment Analysis
To identify differential alternative splicing (DAS) events between four groups (BM, UM, M1, and M3), PSI values for each AS event were calculated using the psiPerEvent function of SUPPA2, based on the transcript per million (TPM) values and the corresponding AS event annotations. Differential splicing analysis was then performed using the diffSplice function in SUPPA2, with statistical significance defined by a threshold of |ΔPSI| ≥ 0.15 and p < 0.05. In parallel, differential expression analysis of transcripts between groups (BM, UM, M1, and M3) was conducted using DESeq2 (v 1.32.0) [45], based on TPM. Differentially expressed transcripts (DETs) were identified using the cutoff |log_2_(fold change)| ≥ 1 and p < 0.05. To explore the functional implications of both significant DAS events and DETs, Gene Ontology (GO) and KEGG pathway enrichment analyses were performed. Gene annotations were first retrieved from eggNOG-mapper results, and enrichment analysis was carried out using clusterProfiler [46]. Only GO terms or KEGG pathways with p < 0.05 were considered significantly enriched.
2.10. Validation of Differentially Expressed Genes
To validate the transcriptome sequencing results, five differentially expressed genes were selected for quantitative real-time PCR (qRT-PCR). Specific primers for the selected five genes were designed using Primer Premier 5.0. To validate differential gene expression between (BM, UM, M1, and M3), two qPCR kits, including the miRcute Plus miRNA qPCR Kit (SYBR Green) and the Talent qPCR Premix (SYBR Green) (both from TIANGEN Biotech, Beijing, China), were used to perform qRT-PCR for the five selected genes based on a LightCycler^®^ 480 system (Roche Applied Science, Indianapolis, IN, USA). 18S rRNA served as the internal control (reference gene). Each gene was amplified in three biological replicates and three technical replicates. Relative expression levels were calculated using the 2^−ΔΔCt^ method [47]. Statistical significance was determined using a Student’s t-test (p < 0.05) implemented in R software (v 4.1.0).
3. Results
3.1. Summary of PacBio Iso-Seq Data and Collapsing Redundant Isoforms
In the present study, PacBio SMRT RNA-seq was performed on RNA samples obtained from four experimental groups (BM, UM, M1, and M3), generating a total of 59,566,665 subreads (approximately 34.0 GB) and 880,044 circular consensus sequences (CCSs).
Following IsoSeq3 refinement, clustering, and polishing, a total of 59,156 high-quality isoforms (average length: 2074.8 bp) and 36 low-quality isoforms (average length: 2293.1 bp) were obtained. Given their minimal proportion, the low-quality isoforms were excluded from subsequent analyses (Table 2). Reference-guided collapsing of redundant transcripts yielded 50,170 unique isoforms. Additionally, pseudo-reference genome construction identified 1483 novel isoforms. The final integration of isoforms using CD-HIT resulted in a non-redundant transcriptome comprising 51,637 isoforms, with an average length of 2056 bp (Table 3).
3.2. Evaluation of Reconstructed Transcriptomes
In this study, the completeness and structural characteristics of the reconstructed transcriptome were comprehensively evaluated (Figure 1). Using BUSCO analysis, we identified 25.8% complete and single-copy orthologs (n = 261), 57.7% complete and duplicated (n = 584), 1.9% fragmented (n = 20), and 14.6% missing orthologs (n = 148) (Figure 1A), indicating a reasonably complete representation of conserved gene content. Comparative analysis with the reference genome annotation revealed that the reconstructed transcriptome comprised 11,101 transcripts matching known isoforms (category “c”), 14,824 potentially novel isoforms sharing splice junctions with known genes (category “j”), and 6289 entirely novel transcripts from previously unannotated loci (category “u”) (Figure 1B). These findings suggest that the reconstructed transcriptome not only recovers a substantial proportion of the reference annotation but also uncovers a considerable number of novel and potentially functionally relevant isoforms.
3.3. Functional Annotation
To gain biological insight into the reconstructed transcriptome, functional annotation was performed and aligned with multiple well-established databases. The number of transcript isoforms annotated varied across different databases, ranging from 26,827 (Swiss-Prot) to 37,883 (TrEMBL), demonstrating broad but variable annotation coverage (Figure 2A). The overlap in annotation results across four databases—Swiss-Prot, TrEMBL, UniRef90, and NR. A total of 38,124 transcript isoforms were annotated in at least one of these databases, among which 26,826 isoforms were commonly identified by all four, reflecting a high degree of annotation consistency and supporting the reliability of the reconstructed transcriptome (Figure 2B). To further characterize transcript functions, isoforms were classified into Clusters of Orthologous Groups (COG) categories, as presented in Figure 2C. The largest group of transcripts was assigned to Category S (Function unknown), followed by Category T (Signal transduction mechanisms) and Category O (Posttranslational modification, protein turnover, chaperones). This functional distribution suggests the transcriptome encompasses a wide range of cellular roles, with a substantial portion representing uncharacterized or potentially novel functions.
3.4. Alternative Splicing Events
To further characterize transcriptomic complexity, we investigated alternative splicing (AS) events in the reconstructed transcriptome. A total of seven distinct AS event types were identified: alternative 3′ splice site (A3), alternative 5′ splice site (A5), alternative first exon (AF), alternative last exon (AL), mutually exclusive exon (MX), retained intron (RI), and skipped exon (SE). A detailed analysis of AS patterns in ovarian tissue revealed that AF (alternative first exon) was the most prevalent event, followed by A5, A3, AL, RI, SE, and MX (Figure 3A; Table 4 and Table S1). To elucidate the biological significance of these splicing events, Gene Ontology (GO) enrichment analysis was performed on all genes undergoing alternative splicing. As shown in Figure 3B, the top ten significantly enriched biological process (BP) terms were predominantly associated with protein regulation and chromatin remodeling. These included peptidyl-serine dephosphorylation, positive regulation of histone deacetylation, and histone monoubiquitination, suggesting that alternative splicing in ovarian tissue may contribute substantially to post-translational modification and epigenetic control mechanisms.
3.5. Differential Alternative Splicing (DAS) Events, Differential Expressed Transcripts (DETs), and Their Differential Expression Analysis
To better understand transcriptomic differences between experimental groups, we examined both differential alternative splicing (DAS) events and differentially expressed transcripts (DETs) across the BM, UM, M1, and M3 samples. The DAS analysis revealed that the number of significant splicing events varied among the six pairwise comparisons. Most of these events were linked to protein-coding genes and were dominated by four splicing types: retained intron (RI), alternative 5′ splice site (A5), alternative 3′ splice site (A3), and alternative first exon (AF) (Table 5; Figure 4A; Table S2). In terms of differential expression, the UM vs. M3 comparison showed the largest number of significantly different transcript isoforms (1963), while M1 vs. M3 had the fewest (921) (Table 5; Figure 4B; Table S3). Interestingly, the most highly differentially expressed transcripts (based on p-value) varied depending on the comparison group. For instance, in the UM vs. M1 group, notable transcripts included PB.3431.2, PB.10089.7 (with domains found in myosin and kinesin tails), PB.10152.5, PB.552.5 (a member of the Rho family of small GTPases), and PB.9323.2. In the UM vs. M3 group, top transcripts included PB.8738.1 (belonging to the uncharacterized UPF0029 protein family), along with PB.5936.9, PB.229.2, PB.9078.1, and PB.9829.3. For the M1 vs. M3 comparison, prominent transcripts included PB.5147.8 (containing a BRIX domain), PB.6586.23 (with a VWC_def domain), PB.2909.2 (from the glycosyl hydrolase family 31), PB.6759.12 (from the class-I aminoacyl-tRNA synthetase family), and PB.705.6 (a DnaJ homolog subfamily B member).
3.6. DAS and DETs Enrichment Analysis
To gain deeper insights into the molecular mechanisms underlying transcriptomic changes, both Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed using genes associated with differentially expressed transcripts (DETs) and differentially alternative splicing (DAS) events. In the UM vs. M1 comparison, the top ten significantly enriched GO biological process terms were primarily linked to signaling pathway activation and neurochemical processes. These included primary amino compound metabolic processes, serotonin metabolism, positive regulation of catecholamine secretion, and negative regulation of cardiac contractility via chemical signaling (Figure 5A).
KEGG pathway analysis further revealed enrichment in biosynthesis and metabolic pathways, such as betalain biosynthesis, isoquinoline alkaloid biosynthesis, indole alkaloid biosynthesis, arachidonic acid metabolism, phenylalanine metabolism, and pyruvate metabolism (Figure 5B). In the UM vs. M3 comparison, GO enrichment pointed predominantly to pathways involved in energy metabolism, including oxidative phosphorylation, ribonucleoside triphosphate biosynthetic process, ATP biosynthetic process, respiratory electron transport chain, and mitochondrial ATP synthesis coupled with electron transport (Figure 5C). The KEGG results highlighted oxidative phosphorylation, non-alcoholic fatty liver disease (NAFLD), and carbon fixation in photosynthetic organisms as the most enriched pathways (Figure 5D). A broader integrative analysis of DAS events and DETs across all six group comparisons revealed a recurring enrichment in functional categories related to cellular energy production, protein complex formation, detoxification, and developmental regulation. The top GO terms included protein tetramerization, oxidative phosphorylation, cellular detoxification, biogenic amine metabolism, glucose catabolic process, and regulation of endodermal cell differentiation (Figure 5E). Correspondingly, KEGG pathway enrichment across comparisons indicated consistent involvement in pathways such as ECM–receptor interaction, oxidative phosphorylation, glycosaminoglycan binding, complement and coagulation cascades, protein digestion and absorption, phenylalanine metabolism, tyrosine metabolism, maturity-onset diabetes of the young, arginine biosynthesis, and isoquinoline alkaloid biosynthesis (Figure 5F). These findings collectively underscore the interplay between transcriptomic regulation, metabolic activity, and tissue-specific functional remodeling.
3.7. Validation of Significant Differential Expression Transcripts
To ensure the accuracy of the transcriptome profiling results, we conducted quantitative real-time PCR (qRT-PCR) validation on a subset of differentially expressed genes (Table S4). Five genes—PB.10563, PB.6079, PB.10649, PB.2978, and PB.3758—were randomly selected for this purpose. The expression patterns obtained from qRT-PCR closely paralleled those from the RNA-seq analysis across the different group comparisons (Figure 6). Specifically, both approaches confirmed that PB.10563, PB.6079, PB.10649, and PB.2978 were significantly upregulated, while PB.3758 was consistently downregulated. These findings confirm the reliability and consistency of the transcriptomic data used in this study.
4. Discussion
Pubertal molting marks a critical transition in the reproductive cycle of crustaceans, signaling the onset of ovarian development. In the mud crabs, mating is known to accelerate this process dramatically [32,33,48]. Yet, despite its biological importance, the molecular landscape underlying this transition has remained largely unexplored. In this study, we applied full-length transcriptome sequencing to comprehensively map the changes in gene expression and alternative splicing (AS) following mating. Our results offer new insights into the transcriptional profile and signaling pathways that drive post-mating ovarian maturation.
The results of this study indicate that mating exerts a significant impact on multiple molecular processes in the ovary, particularly affecting cell signal transduction, neurochemical pathways, and metabolic networks. These findings suggest that mating may act as a key external stimulus to initially activate ovarian development–related signaling pathways, thereby regulating the supply of materials needed for oocyte growth and subsequent yolk accumulation. Further comparison between the unmated and three days post-mating (UM vs. M3) ovaries revealed substantial remodeling of energy metabolism, especially mitochondrial function and ATP production, reflecting the increased energy demand during rapid ovarian development. In addition, enrichment of the NAFLD pathway suggests potential lipid metabolic dysregulation, possibly associated with fatty acid accumulation. Collectively, these results indicate that mating not only initiates ovarian development through neuroendocrine signaling but also supports oocyte growth and maturation by reorganizing energy and lipid metabolism.
A high-quality transcriptome is essential for investigating dynamic developmental processes like oogenesis. The BUSCO completeness of the reconstructed transcriptome was 25.8%, which was significantly lower than that of the reference genome [36]. This difference likely reflects the tissue-specific and temporally restricted nature of ovarian gene expression [49,50]. Encouragingly, the proportion of fragmented transcripts in our dataset was only 1.9%, which is lower than a previous SMRT-sequencing-based transcriptome (3.2%) [51], highlighting the reliability and accuracy of the sequencing and data processing. The reconstructed transcriptome also revealed substantial isoform diversity, including over 26,000 transcripts not previously annotated—many of which may be novel regulators of ovarian development.
Alternative splicing analysis revealed extensive transcript diversity, with the most frequent AS events being alternative first exon (AF) and alternative 5′ splice site (A5). These splicing patterns were enriched in genes associated with chromatin remodeling, protein regulation, and metabolic processes, suggesting a shift in cellular priorities to support oocyte maturation. It is well known that histone deacetylation inhibits gene expression by removing acetyl groups from active genes. In mice, histone deacetylation occurs rapidly following ovulatory signal induction, indicating its crucial role in ovarian development [52,53,54]. Monoubiquitination of histones is closely associated with the transcriptional regulation of gene expression and the DNA damage response [55]. The presence of such complex isoform architecture implies that AS plays a fundamental role in fine-tuning gene function during ovarian development [56].
Through differential expression and AS analyses, we observed significant transcriptional shifts across developmental stages. The highest number of differentially expressed transcripts (1963) was found in the UM vs. M3 comparison, highlighting how mating triggers a profound molecular reprogramming in the ovary. Several upregulated genes stood out due to their functional importance. For instance, glutathione S-transferase (GST) may reflect increased antioxidant defense during rapid tissue growth [57], ATP synthase supports high energy demands during oocyte proliferation [58,59], and LPIN2-like is likely involved in lipid redistribution and membrane biosynthesis essential for yolk accumulation [58,60,61]. During ovarian development, particularly in the stages of oocyte growth and yolk accumulation, cells required extensive protein synthesis to meet the demands of oocyte development. In the comparative analysis, genes associated with protein synthesis, including EIF3C, EF1a-F2, and EIF3L, were significantly upregulated. This indicates that translation initiation and elongation pathways were markedly activated during the critical stages of ovarian development, thereby enhancing protein synthesis efficiency and providing molecular support for the rapid growth and yolk accumulation of oocytes. These findings further suggested that the regulation of protein synthesis represents one of the central mechanisms underlying ovarian development in crabs.
Pathway enrichment analyses offered even deeper insights. In the UM vs. M1 comparison, GO enrichment pointed to the activation of neurochemical signaling pathways, especially those involving serotonin and catecholamines. These amino acid-derived compounds are known to regulate reproductive hormones in crustaceans and fish alike [62]. For example, serotonin stimulates gonadotropin release in the thoracic ganglia [63], while tyrosine-derived metabolites like dopamine and N-acetylserotonin fluctuate with ovarian stage in Chinese sturgeon, peaking during active vitellogenesis [64]. Among catecholamines, dopamine has been the most extensively studied. During ovarian development, dopamine concentrations generally exhibited a decreasing trend, and numerous studies have confirmed its inhibitory role in oocyte maturation and ovulation [65,66,67]. In contrast, serotonin plays a crucial stimulatory role in regulating oocyte maturation [68]. It is therefore speculated that serotonin and catecholamine signaling pathways may jointly contribute to the neuroendocrine regulation of ovarian development in crabs, and their dynamic changes at different developmental stages may form an “inhibition–activation” molecular switch mechanism that finely regulates the progression of oocyte development. The observed enrichment of these pathways in our data suggests that serotonin and catecholamine signaling may be among the earliest molecular responses to mating, serving as upstream activators of reproductive gene networks [69]. KEGG enrichment analysis showed that mating significantly affected cell signal transduction, neurochemical processes, and metabolic pathways. These changes suggest that mating may initially activate certain ovarian development–related signaling pathways, thereby regulating the provision of materials required for subsequent ovarian development.
In contrast, the UM vs. M3 comparison highlighted enrichment of energy metabolism-related pathways, particularly those associated with mitochondrial function and ATP production, including oxidative phosphorylation, ATP biosynthesis, and electron transport chain activity. These pathways reflect the high energy demands associated with vitellogenesis and oocyte growth [70]. Supporting this, KEGG analysis revealed significant enrichment in pathways like pyruvate metabolism, phenylalanine metabolism, and even non-alcoholic fatty liver disease—systems that are increasingly recognized for their roles in nutrient sensing and energy regulation during reproduction [71]. During ovarian development in Eriocheir sinensis, phospholipid metabolism and fatty acid transport pathways were markedly upregulated, indicating a high energy demand for ovarian maturation [72]. In addition, the enrichment of NAFLD suggested a potential metabolic dysregulation in the ovary, which may be related to fatty acid accumulation. This post-mating shift toward bioenergetic activation is consistent with observations in Portunus trituberculatus, where energy metabolism genes are downregulated in later stages, likely indicating a transition from energy storage to energy utilization [73].
An intriguing finding was the significant enhancement of host photosynthetic carbon fixation pathways observed three days after mating (Figure 5D). Although the present study did not directly measure changes in the microbiome, a plausible hypothesis is that this phenomenon may be associated with microbial community restructuring following the mating–molting cycle. In S. paramamosain, mating typically occurs shortly after female molting. During molting, the old exoskeleton and its associated microbial communities are shed, creating a “blank slate” for the colonization of new microbes [74]. We speculate that this secondary succession of the microbiome may recruit photosynthetic bacteria capable of carbon fixation. The carbon fixed by these symbionts could be directly assimilated by the host to support energetically demanding reproductive activities, such as vitellogenesis and embryonic development. This hypothesis proposes a previously unrecognized microbiome-based nutritional symbiosis strategy, linking molting, reproduction, and energy metabolism. Future studies measuring microbiome changes before and after molting and mating, combined with stable isotope tracing, may help to test this hypothesis.
Structural remodeling of the ovary also emerged as a central theme. The extracellular matrix (ECM)-receptor interaction pathway, enriched in our analysis, is critical for follicular development and oocyte differentiation [75]. In crustaceans and other animals, ECM components such as laminin and collagen interact with integrin receptors to mediate key processes like steroidogenesis, cell migration, and structural integrity [76,77]. These interactions activate intracellular pathways—including TGF-β and PI3K-Akt—that orchestrate follicular growth and survival [78,79]. Given the enrichment of these pathways in our dataset, it’s likely that ECM remodeling contributes to the physical restructuring of the ovary required for oocyte maturation. We also observed enrichment in the HIF-1 and IL-17 signaling pathways. HIF-1 is known to regulate oxygen availability in growing tissues and has been implicated in modulating oocyte developmental timing and blastocyst viability [80,81]. The IL-17 pathway, though traditionally associated with immune function, has recently been linked to follicular maturation in eels [82,83], and may serve a similar role in S. paramamosain by regulating ovarian inflammation and tissue remodeling during maturation.
Lipids are vital during ovarian development, not just as structural components but as key energy reserves for embryos and larvae. In Scylla olivacea, fatty acids such as palmitic acid increase significantly during vitellogenesis [84], supporting the hypothesis that lipid mobilization supports later-stage oocyte development. In our study, enrichment of pathways like arachidonic acid metabolism and nitrogen metabolism suggests that similar mechanisms are at play in S. paramamosain. The upregulation of LPIN2-like, known to regulate phospholipid and triglyceride synthesis, further supports the role of lipid remodeling during this critical phase.
Finally, oxidative stress management appears essential for successful oogenesis. While reactive oxygen species (ROS) can serve as signaling molecules that promote oocyte maturation [85,86], their overaccumulation can cause cellular damage. Our data revealed strong enrichment in GO terms related to cellular oxidant detoxification, with genes such as SOD2, CAT, GPX4, and GSTO1 being significantly upregulated. These enzymes form the core of the antioxidant defense system and likely help maintain redox balance in the rapidly developing ovarian tissue [87,88]. Reduced antioxidant expression in other species has been linked to disrupted ROS balance and altered oocyte development, suggesting that proper ROS regulation is a conserved feature of reproductive biology.
5. Conclusions
This study provides a full-length transcriptomic framework for understanding ovarian development following mating in the mud crab (Scylla paramamosain). By integrating differential gene expression and alternative splicing analyses, we found that mating initiates extensive transcriptional reprogramming in the ovary. Early responses were characterized by the activation of serotonin and catecholamine signaling pathways, highlighting their potential roles as upstream regulators of reproductive hormones. As ovarian development progressed, pathways related to oxidative phosphorylation, ATP production, lipid metabolism, and extracellular matrix remodeling became dominant, reflecting the increased energy and structural material requirements of vitellogenesis and oocyte maturation. In addition, the upregulation of antioxidant defense genes suggests that maintaining redox homeostasis is crucial for successful oogenesis. Together, these findings not only deepen our understanding of the molecular regulation of reproduction in S. paramamosain but also provide valuable insights that may inform broodstock management and reproductive control strategies in aquaculture.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sanda T. Shimizu T. Iwasaki T. Dan S. Hamasaki K. Effect of Temperature on Survival, Intermolt Period, and Growth of Juveniles of Two Mud Crab Species, Scylla paramamosain and Scylla serrata (Decapoda: Brachyura: Portunidae), under Laboratory Conditions Nauplius 202230 e 202201210.1590/2358-2936 e 2022012 · doi ↗
- 2Zhao Y. Waqas W. Cui W. Ye S. Gao W. Zhang Q. Lin Z. Zhu D. Lin F. Ikhwanuddin M. Comparative Analysis of Embryonic Development and Growth Performance among Two Mud Crab Species and Their Hybrids Aquaculture 202559674179510.1016/j.aquaculture.2024.741795 · doi ↗
- 3Ye H. Tao Y. Wang G. Lin Q. Chen X. Li S. Experimental Nursery Culture of the Mud Crab Scylla paramamosain (Estampador) in China Aquac. Int.20111931332110.1007/s 10499-010-9399-3 · doi ↗
- 4Nghia T.T. Wille M. Binh T.C. Thanh H.P. Van Danh N. Sorgeloos P. Improved Techniques for Rearing Mud Crab Scylla Paramamosain (Estampador 1949) Larvae Aquac. Res.2007381539155310.1111/j.1365-2109.2007.01814.x · doi ↗
- 5Fishery Bureau of Ministry of Agriculture of China China Fishery Statistical Yearbook 2024 Chinese Agricultural Press Beijing, China 2024
- 6Quinitio E.T. De Pedro J. Parado-Estepa F.D. Ovarian Maturation Stages of the Mud Crab Scylla serrata Aquac. Res.2007381434144110.1111/j.1365-2109.2007.01650.x · doi ↗
- 7Christy J.H. Timing of Hatching and Release of Larvae by Brachyuran Crabs: Patterns, Adaptive Significance and Control Integr. Comp. Biol.201151627210.1093/icb/icr 01321593141 · doi ↗ · pubmed ↗
- 8Waiho K. Fazhan H. Shahreza M.S. Moh J.H.Z. Noorbaiduri S. Wong L.L. Sinnasamy S. Ikhwanuddin M. Transcriptome Analysis and Differential Gene Expression on the Testis of Orange Mud Crab, Scylla olivacea, during Sexual Maturation P Lo S ONE 201712 e 017109510.1371/journal.pone.017109528135340 PMC 5279790 · doi ↗ · pubmed ↗