oxLDL-induced aortic valve inflammation and calcification: opportunities for clinical translation
Yu-Jen Wang, Michael A Matter, Christian M Matter

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
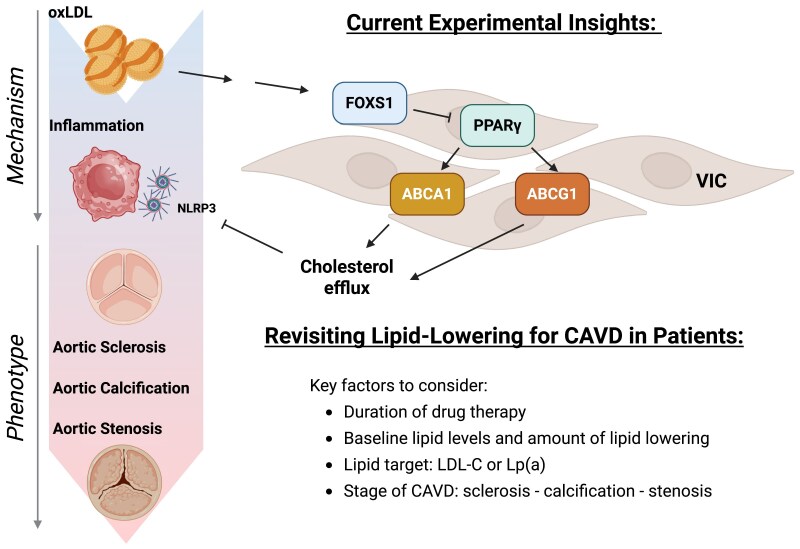 Figure 1
Figure 1- —Swiss National Science Foundation10.13039/501100001711
- —Swiss Heart Foundation10.13039/501100004362
- —Lindenberg Family Office
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInfective Endocarditis Diagnosis and Management · Aortic Thrombus and Embolism · Cardiac tumors and thrombi
This editorial refers to ‘OxLDL-induced FOXS1 mediates cholesterol transport dysfunction and inflammasome activation to drive aortic valve calcification’, by C. Jiang et al., https://doi.org/10.1093/cvr/cvaf159.
Calcific aortic valve disease—lack of drug treatment
Calcific aortic valve disease (CAVD) represents one of the most prevalent cardiovascular diseases in the aging population.^1^ It can progress to relevant aortic stenosis and accounts for significant cardiac morbidity and mortality. Current European guidelines on the treatment of aortic valve stenosis^2^ focus on surgical or interventional treatment modalities that comprise valvular repair or replacement. Although very effective, those modalities confer obvious risk and morbidity given their invasiveness. Additionally, they are directed against clinically, functionally and/or morphologically advanced CAVD.
To date, there is no approved drug treatment for CAVD. The recommendations in the current guidelines are limited to treatment of cardiovascular comorbidities.^3^ Notably, the role of lipid-lowering agents, in particular statins, is explicitly negated based on the negative results from two Phase 3 trials testing the effects of high-intensity statin on the progression of aortic valve stenosis.^4,5^ This leaves the question open, whether dysfunctional lipid metabolism is relevant in the early phases of aortic valve stenosis. The current study by Jiang et al.^6^ offers interesting new mechanistic insights, rekindling the spark of interest in potential drug treatment for CAVD (Figure 1).
Dyslipidaemia and inflammation are critical contributors to experimental aortic valve calcification and subsequent stenosis. Currently, no pharmacological therapy is available to delay the progression of aortic valve disease in patients. Previous lipid-lowering trials failed to demonstrate clinical benefit. Jiang et al. now report that oxLDL induces FOXS1 expression in VIC, which suppresses PPARγ activity, impairs cholesterol efflux (via ABCA1/ABCG1), and activates the NLRP3 inflammasome. This novel mechanistic insight prompts a re-evaluation of lipid-lowering strategies in patients with aortic sclerosis up to calcific aortic valve stenosis. In particular, factors such as duration of drug therapy, baseline lipid levels and dosing regimens of lipid-lowering therapies, the targeted lipid species (e.g. LDL-C and Lp(a)), and aortic valve disease stage likely influence the therapeutic effects. oxLDL, oxidized low-density lipoprotein; FOXS1, forkhead box S1; PPARγ, peroxisome proliferator-activated receptor γ; ABCA1, ATP binding cassette subfamily A member 1; ABCG1, ATP-binding cassette subfamily G member 1; NLRP3, nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; VIC, valvular interstitial cell; CAVD, calcific aortic valve disease; LDL-C, low-density lipoprotein cholesterol; Lp(a), lipoprotein(a).
oxLDL and inflammation in experimental calcific aortic valve disease
In this issue of Cardiovascular Research, Jiang et al. elucidated the molecular mechanisms by which oxidized low-density lipoprotein (oxLDL) exacerbated aortic valve calcification by activating the transcription factor Forkhead Box S1 (FOXS1). Initially, the authors identified increased expression of FOXS1 in valvular interstitial cells (VICs) of human calcified, stenotic aortic valves compared to normal ones. Ablation of FOXS1 mitigated valve calcification in both ex vivo human VICs and in vivo mouse models. Interestingly, transcriptomic data revealed a correlation between FOXS1 expression and cholesterol transport pathways. Applying knockdown and pharmacological strategies, they demonstrated that FOXS1 regulated the expression of the lipid transporters ATP binding cassette subfamily A member 1 (ABCA1) and ATP-binding cassette subfamily G member 1 (ABCG1), both established downstream targets of peroxisome proliferator activated receptor γ (PPARγ) in VICs. Decreased levels of these cholesterol transporters induced cholesterol accumulation and inflammatory responses (interleukin [IL]-1β, IL6, and NLRP3 [nucleotide-binding domain, leucine-rich-containing family, pyrin domaincontaining-3] inflammasome activation) associated with CAVD. Finally, they identified an upstream regulator BSCL2 (lipid droplet biogenesis associated, seipin) of PPARγ, highlighting its potential to treat CAVD by promoting cholesterol efflux.
Both dysregulation of lipid metabolism and inflammation play a role in atherosclerotic cardiovascular disease (ASCVD).^7^ However, the mechanisms underlying CAVD related to lipid dysregulation and inflammation are much less understood.3 Although differences are present, similar key concepts underlying the pathophysiology of atherosclerosis and CAVD may be presumed. Along this line, in vitro oxLDL stimulation and in vivo atherosclerosis-prone mouse models serve as important experimental tools for investigating molecular mechanisms of CAVD. Moreover, a proteomic-based comparison between human calcified atherosclerotic plaques and human calcified aortic valves revealed shared lipoprotein-related pathways between the two diseases, underscoring an association of lipid dysregulation with CAVD.^8^
Translating experimental insights of CAVD to the clinical context
Far less is known about the value of targeting lipid dysregulation and/or inflammation in CAVD than in ASCVD. An indicator towards a potential causal role of dyslipidaemia in progression of CAVD is presented by a study of subjects with heterozygous familial hypercholesterolemia, for whom an association between increased LDL–cholesterol and CAVD has been reported.^9^ Furthermore, genetic variation in the lipoprotein(a) [Lp(a)] locus, mediated by Lp(a) levels, has been identified as a causal factor for patients with aortic valve calcification.^10^ Lp(a) induced increased calcium deposition and the expression of pro-inflammatory cytokines.^11^ These findings suggest a causal role of LDL–cholesterol and Lp(a) for CAVD.
The present study offers interesting food for thought towards clinical translation (Figure 1). As mentioned above, several large-scale randomized controlled trials have shown no benefit of lipid-lowering treatment to prevent progression of aortic stenosis. However, those studies presented only relatively short-term median follow-up periods of 25 or 52.2 months, respectively.^4,5^ In terms of echocardiographic measures, CAVD typically progresses at an average rate of an increase in aortic velocity of about 0.16 m/s per year, and a decrease in valve area of 0.08 cm² per year.^12^ Once reaching a transvalvular aortic velocity of 2 m/s or more, progression to severe aortic stenosis typically occurs within 10 years. Thus, the time required to study a potential treatment effect of lipid-lowering therapies to prevent or decelerate progression of CAVD may be far in excess of what previous studies have investigated. Moreover, the magnitude of effects of lifelong genetic lipid changes on CAVD have to be placed in perspective to the duration of drug-based lipid-lowering.
Additionally, previous clinical studies in this field described a setting where at least mild CAVD is already present, i.e. a setting of secondary prevention. The current study by Jiang et al. demonstrates a role of lipid dysregulation and subsequent inflammation at the initiation and very early stages of CAVD. Translated into clinical application, the use of lipid-lowering or anti-inflammatory agents to prevent the onset or progression of CAVD in a primary prevention setting remains to be explored for LDL-C and inflammation (Figure 1)—or is being investigated for Lp (a) (NCT 05646381).
Extending the notions thus put forward beyond traditional markers of lipid dysregulation, there has been a recent surge in interest in the causal role of Lp(a) in both ASCVD and CAVD, as already mentioned above.^13^ Ongoing studies targeting Lp(a) promise important new insights and potentially new treatment options for both ASCVD and CAVD.
Conclusion and perspectives
New mechanistic insights into the pathophysiological processes underlying the initiation and progression of CAVD may unravel novel drug targets (Figure 1). The results put forward by Jiang et al. reignite the spark of recently dwindling interest in traditional lipid-lowering drugs to treat or prevent CAVD. Jiang et al. also provide fuel for thought on potential novel therapeutic targets, linking lipid dysregulation and inflammation, a concept which may prove fruitful within as well as beyond the setting of CAVD.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Moncla LM, Briend M, Bosse Y, Mathieu P. Calcific aortic valve disease: mechanisms, prevention and treatment. Nat Rev Cardiol 2023;20:546–559.36829083 10.1038/s 41569-023-00845-7 · doi ↗ · pubmed ↗
- 2Vahanian A, Beyersdorf F, Praz F, Milojevic M, Baldus S, Bauersachs J, Capodanno D, Conradi L, De Bonis M, De Paulis R, Delgado V, Freemantle N, Gilard M, Haugaa KH, Jeppsson A, Juni P, Pierard L, Prendergast BD, Sadaba JR, Tribouilloy C, Wojakowski W. 2021 ESC/EACTS guidelines for the management of valvular heart disease; ESC/EACTS Scientific Document Group. Eur Heart J 2022;43:561–632.34453165 10.1093/eurheartj/ehab 395 · doi ↗ · pubmed ↗
- 3Blaser MC, Back M, Luscher TF, Aikawa E. Calcific aortic stenosis: omics-based target discovery and therapy development. Eur Heart J 2025;46:620–634.39656785 10.1093/eurheartj/ehae 829PMC 11825147 · doi ↗ · pubmed ↗
- 4Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer R; SEAS Investigators. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med 2008;359:1343–1356.18765433 10.1056/NEJ Moa 0804602 · doi ↗ · pubmed ↗
- 5Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA; Scottish Aortic Stenosis and Lipid Lowering Trial, Impact on Regression (SALTIRE) Investigators. A randomized trial of intensive lipidlowering therapy in calcific aortic stenosis. N Engl J Med 2005;352:2389–2397.15944423 10.1056/NEJ Moa 043876 · doi ↗ · pubmed ↗
- 6Jiang C, Yao D, Shen Q, Tian R, Fan L, Zheng Q, Qian X, Liu Z, Huang Y, Dong N. Oxidized LDL-induced FOXS 1 mediates cholesterol transport dysfunction and inflammasome activation to drive aortic valve calcification. Cardiovasc Res. 2025;121:1941–1955.10.1093/cvr/cvaf 159PMC 1255139140990096 · doi ↗ · pubmed ↗
- 7Libby P, Ridker PM, Hansson GK; Leducq Transatlantic Network on Atherothrombosis. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol 2009;54:2129–2138.19942084 10.1016/j.jacc.2009.09.009PMC 2834169 · doi ↗ · pubmed ↗
- 8Blaser MC, Buffolo F, Halu A, Turner ME, Schlotter F, Higashi H, Pantano L, Clift CL, Saddic LA, Atkins SK, Rogers MA, Pham T, Vromman A, Shvartz E, Sukhova GK, Monticone S, Camussi G, Robson SC, Body SC, Muehlschlegel JD, Singh SA, Aikawa M, Aikawa E. Multiomics of tissue extracellular vesicles identifies unique modulators of atherosclerosis and calcific aortic valve stenosis. Circulation 2023;148:661–678.37427430 10.1161/CIRCULATIONAHA.122.063402 PMC 10527599 · doi ↗ · pubmed ↗