Synthesis of Sterically Congested Carbonyl Compounds via an ipso-Selective Sulfonium Rearrangement
Immo Klose, Christian Knittl-Frank, Nicolas G.-Simonian, Boris Maryasin, Daniel Kaiser, Nuno Maulide

TL;DR
A new method is introduced for making crowded carbonyl compounds using a sulfonium rearrangement, which works well where traditional methods fail.
Contribution
The ipso-selective sulfonium rearrangement provides a novel, metal-free route for synthesizing sterically congested α-arylated carbonyl compounds.
Findings
The method enables synthesis of α-arylated carbonyl compounds that are difficult to make via traditional cross-coupling.
The sulfonium-based rearrangement is robust against steric hindrance.
This approach is orthogonal to metal-catalyzed strategies.
Abstract
Despite the success of metal-catalyzed cross-coupling strategies to assemble a variety of C–C bonds, the synthesis of sterically congested α-arylated carbonyl compounds remains a difficult task. Herein, we present an ipso-rearrangement approach relying on electron redistribution in sulfonium intermediates. This modular method enables the synthesis of a variety of α-arylated carbonyl compounds and is orthogonal to traditional metal-catalyzed cross-coupling strategies. More importantly, it demonstrated remarkable robustness toward steric hindrance and allowed us to efficiently forge congested C–C bonds.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
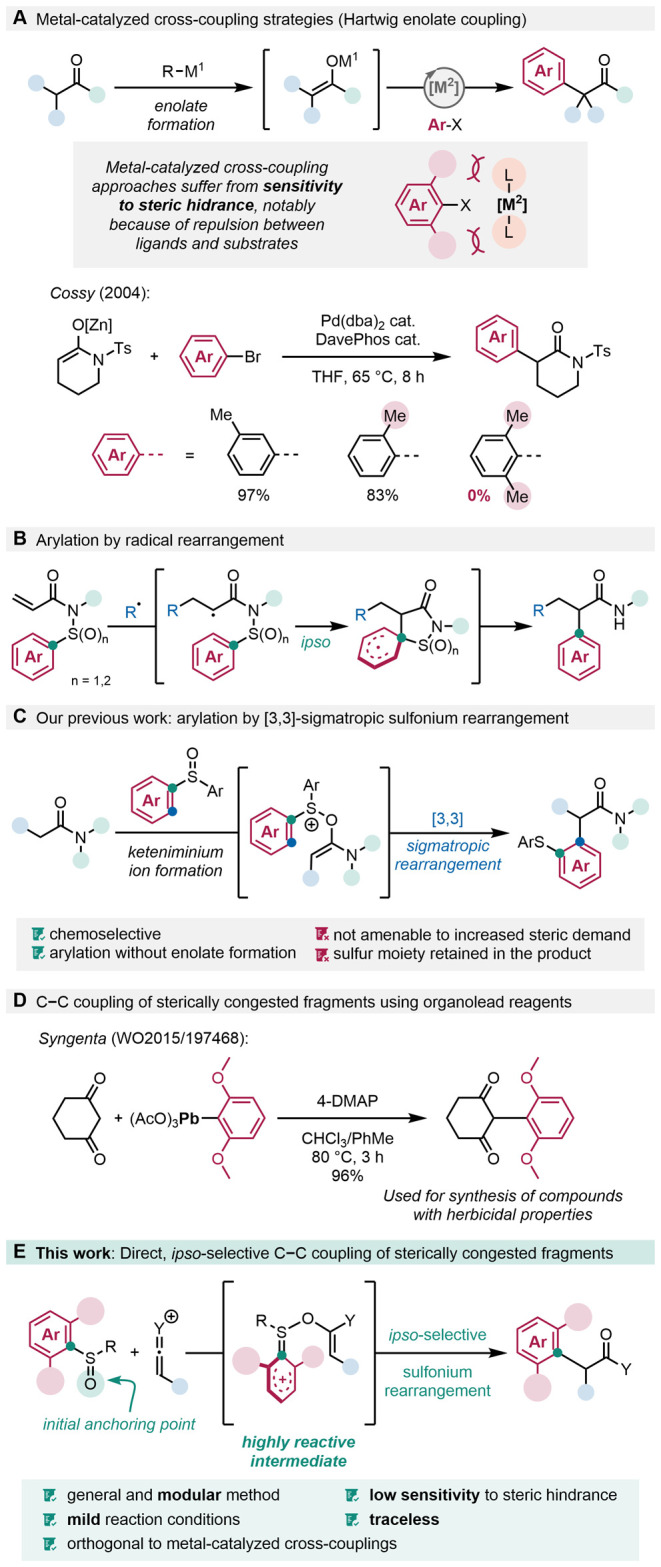 Figure 1
Figure 1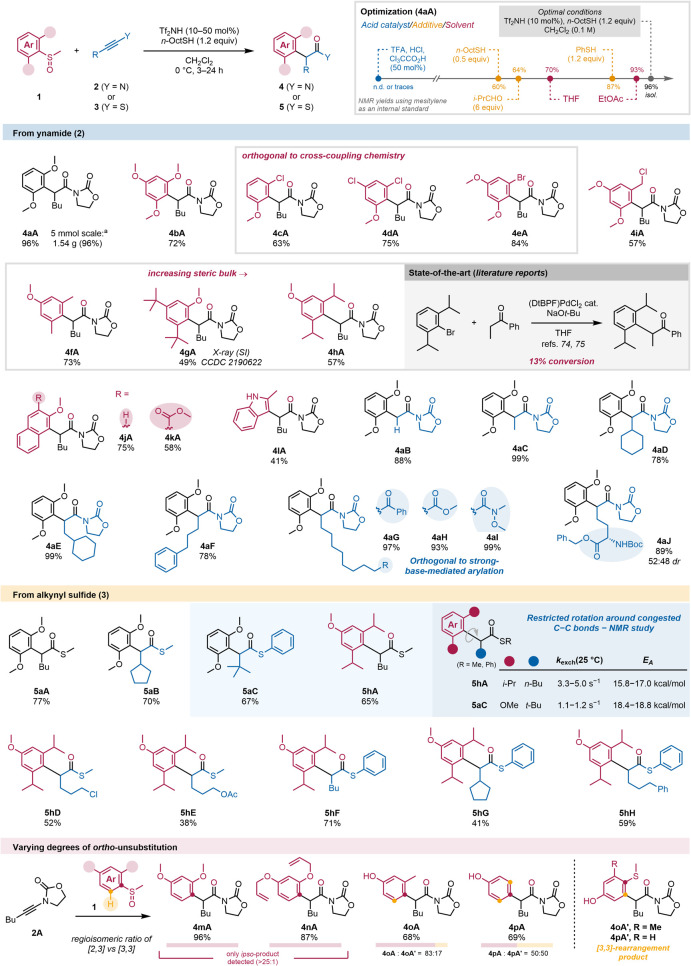 Figure 2
Figure 2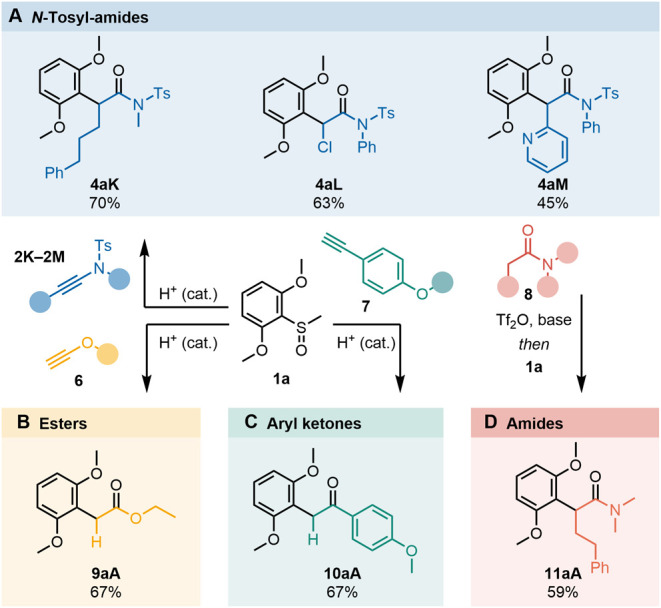 Figure 3
Figure 3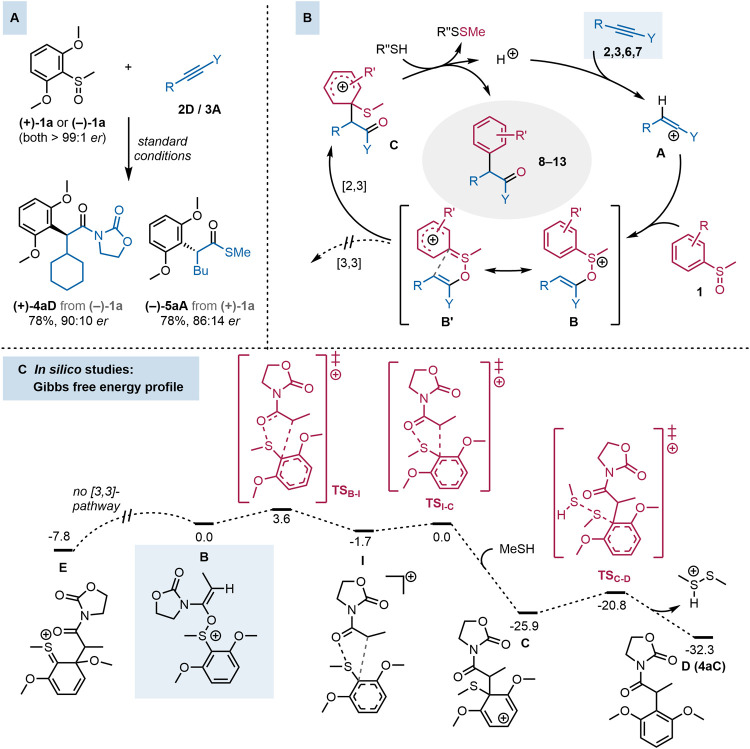 Figure 4
Figure 4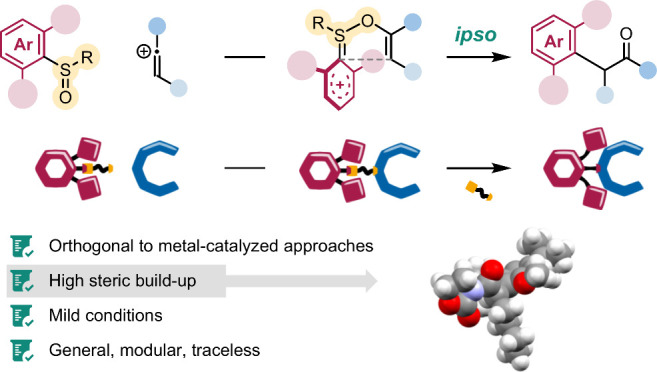 Figure 5
Figure 5- —H2020 European Research Council10.13039/100010663
- —H2020 European Research Council10.13039/100010663
- —H2020 European Research Council10.13039/100010663
- —?sterreichischen Akademie der Wissenschaften10.13039/501100001822
- —Austrian Science Fund10.13039/501100002428
- —Austrian Science Fund10.13039/501100002428
- —Universit?t Wien10.13039/501100003065
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSulfur-Based Synthesis Techniques · Catalytic C–H Functionalization Methods · Synthesis and Catalytic Reactions
Carbonyl compounds bearing an aromatic substituent at the α-position are often of biological relevance, and developing methods for their efficient preparation has been a focal point of contemporary synthesis.? The search for methods allowing for the formation of α-arylated carbonyl compounds naturally invoked metal-catalyzed approaches, perhaps the most notable of which are the Hartwig enolate coupling ?−? ? ? ? ? as well as polarity-reversed α-arylations (FigureA). ?−? ? Unfortunately, while transition metal-catalyzed cross-coupling has become a workhorse in organic synthesis, allowing for the assembly of a variety of C–C bonds, its classical oxidative addition/reductive elimination logic means that metal complexestypically carrying already bulky ligandsface considerable challenges when attempting to couple precursors that are themselves sterically demanding. ?,?−? ? ? ?
On the other hand, the recent renaissance of radical chemistry and photoredox catalysis has shown that open-shell intermediates are less sensitive to the constraints of steric hindrance. ?−? ? ? ? ? ? ? However, control of the reactivity of these highly energetic species can be challenging. This is often addressed by using one reactant in a large excess, ?−? ? or by rendering the process intramolecular, which in turn reintroduces the problem of steric hindrance (FigureB). ?−? ? ? ? ?
Throughout the past decade, Yorimitsu, Procter, ourselves and others have demonstrated the ability of sulfonium rearrangements to form highly congested C–C bonds between sp^3^-hybridized tertiary-quaternary centers or sp^2^–sp^3^-hybridized aryl-tertiary centers (FigureC). ?−? ? ? ? ? ? ? We thus became intrigued by the prospect of developing a sulfonium rearrangement of sp^2^-hybridized aryl sulfoxides capable of tolerating an increased steric demand in the context of a formal carbonyl arylation transformation.
Herein, we present a C–C coupling reaction for the formation of α-aryl carbonyl compounds through an unusual ipso-type rearrangement of a sulfonium intermediate arising from the reaction between an aryl sulfoxide and a vinyl cation (FigureE). The sulfoxide’s oxygen atom serves as the initial anchoring point for the two reactants, similar to a native chemical ligation, with this anchoring event giving rise to a highly reactive sulfonium intermediate, with a notable driving force toward rearrangement. The redirected process toward the ipso-position is related to [2,3]-sigmatropic rearrangements of sulfonium species which have been widely applied for C–heteroatom and C–C bonds formations. ?−? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? This is in contrast to transition metal-catalyzed cross-coupling reactions, as this assembly takes place one atom removed from the point of highest steric congestion, thereby facilitating proximity between the two reactive sites.
The transformation we describe herein is orthogonal to transition metal-catalyzed cross-coupling reactions and offers both traceless? and modular access to previously challenging molecular architectures. It is noteworthy that, despite their seemingly simple structures and well-documented bioactivity, ?−? ? ? these compounds require arduous synthetic routesoften involving stoichiometric amounts of toxic heavy metals such as lead (FigureD). ?,?
During our studies on the reactivity of vinyl cation intermediates with sulfoxides, ?,? we observed that o,o’-disubstituted aryl sulfoxides strayed from the established reactivitywhich would direct C–C bond formation to the ortho-position through [3,3]-sigmatropic rearrangement.
Indeed, initial experiments yielded products of desulfitative coupling to the ipso-position in poor yield (Figure, top-right box). ?−? ? ? As an example, treating an ynamide such as 2A with acid in the presence of sterically hindered aryl sulfoxide 1a, we initially observed the formation of 4aA only in traces. Optimization of this process (see SI for details) provided an increase in yield (96%, even on a gram scale). Arguably the key finding of the optimization process was the realization that the use of a thiol additive completely suppressed undesired side reactions and allowed the use of catalyst loadings as low as 2 mol % (see Figure and Supporting Information (SI)). This screening of conditions also revealed that the reaction is very robust to changes in scale, temperature, solvent, and concentration. Additionally, this transformation can be performed in ethyl acetate (93% NMR yield), an important alternative solvent from the vantage point of green chemistry,? and does not require aqueous workup.?
We first set out to study the scope of sulfoxides and were eager to explore how the method would fare in comparison with transition-metal-catalyzed processes. Pleasingly, we found that highly electron-rich aryl moieties, which are challenging substrates due to their impeded oxidative addition, work efficiently and afford the products in good yields (e.g., 4bA).? Moreover, a variety of halide-substituted sulfoxides were found to be tolerated by this protocol and delivered the corresponding α-arylated products 4cA–4eA (63–84%), further showcasing the orthogonality of this method to conventional metal-catalyzed cross-couplings.
Furthermore, the sensitivity of the reaction toward steric hindrance was assessed by reacting sulfoxides bearing sterically demanding groups at both ortho-positions. Gratifyingly, o,o’-dimethylbenzene-substituted sulfoxide 1f delivered α-arylated carbonyl product 4fA in 73% yield. Remarkably, an aryl sulfoxide bearing two tert-butyl groups still afforded 4gA in 49% yield (see SI for the X-ray structure of 4gA). Even the highly sterically demanding o,o’-diisopropylbenzene-substituted sulfoxide 1h yielded product 4hA in 57% yield. This contrasts sharply with reported methods for preparing an o,o’-diisopropylphenyl-α-arylated carbonyl compound, using Pd-catalysis, which proceeds with poor conversion (13%). ?,?
Functional-group tolerance and the effects of varying substitutions on the electrophilic reaction partner were then explored. Even excellent electrophiles such as benzylic chlorides are well tolerated (4iA, 57% yield), as well as bicyclic structures 4jA and 4kA (75% and 58% yield). An indole sulfoxide could also be coupled, affording product 4lA in a 41% yield. Moreover, a wide range of ynamides (2) with varying substitution pattern could be coupled successfully, forming products 4aB–4aF in high yields (78%–99%). The method also proved to be tolerant of other sensitive functional groups contained within the ynamide substrate, such as a ketone (4aG, 97%), an ester (4aH, 93%) or a Weinreb amide (4aI, 99%). Notably, the synthesis of such products, possessing multiple carbonyl groups, using enolate-based arylation (in the presence of strong bases) would be very challenging due to chemoselectivity issues. Strikingly, despite the acidic reaction conditions, an N-Boc-O-Bn protected amino acid was tolerated under the reaction conditions and provided the corresponding product 4aJ in an excellent yield (89%, 52:48 dr). This result opens an avenue toward the construction of complex amino acid derivatives. Importantly, the successful reaction demonstrates that the threat of intramolecular interception of the highly reactive vinyl cation intermediate can be prevented by careful design of protecting groups.
We next turned our attention to another family of readily available vinyl cation precursors, alkynyl sulfides (3), and found that the reaction between 3A and 1a delivered α-arylated thioester 5aA in 77% yield. Sterically more hindered cyclopentyl- and tert-butyl-substituted alkynyl sulfides 3B and 3C also smoothly underwent this ipso-selective coupling to furnish thioesters 5aB (70%) and 5aC (67%), respectively.
As this indicates that these reaction partners are also competent to form congested C–C bonds, we reacted our o,o’-diisopropylphenyl-substituted sulfoxide with different alkynyl sulfides, obtaining the corresponding thioesters 5hA, 5hD–5hH in yields ranging from 38% to 71%. Remarkably, thioester 5hG, bearing two o,o’-isopropyl groups on the aryl and a secondary β-center, was obtained in a respectable 41% yield.
Such steric congestion could be diagnosed by NMR studies, which showed a relatively slow rotation around the newly formed C–C bond at room temperature. For the o,o’-diisopropylphenyl product 5hA, the exchange constant (k exch,(25 °C)) was measured to be in the region of 4 s^–1^, whereas for α-tert-butyl product 5aC, k _ exch _ was measured to be roughly 1.1 s^–1^ (see SI).
To investigate whether steric or electronic factors drive the ipso-selective rearrangement, we tested several sulfoxides bearing a hydrogen atom in the ortho-position. To our surprise, sulfoxides 1m and 1n exclusively afforded products of ipso-coupling (4mA and 4nA, 96% and 87% yield, respectively) with no observable amounts of the potentially competing products resulting from [3,3]-sigmatropic rearrangement onto the ortho-position (>25:1). This demonstrates that the well-developed ortho-functionalization of aryl sulfoxides can be completely rerouted toward the ipso-coupling by suitable electron distribution of the aryl moiety. However, upon decreasing electron density by replacement or omittance of oxygen substituents, the product of ortho-rearrangement reappeared, leading to mixtures of the two products. Thus, whereas 4oA still showed a preference for ipso-rearrangement (83:17), an aryl sulfoxide bearing a sole p-hydroxy substituent resulted in a 1:1 mixture of [2,3]- and [3,3]-rearrangement products 4pA and 4pA’.
The generality of this coupling is highlighted by the fact that hindered aryl sulfoxides were readily ipso-coupled with a variety of donor-substituted alkynes without further optimization (FigureA–C).
As shown, sulfonamide-derived ynamides (2) afforded the corresponding α-aryl amides 4aK–4aL (FigureA). The reaction was even amenable to a Brønsted-basic pyridine moiety, affording 4aM in 45% yield when using 1.2 equiv of a Brønsted acid. Similarly, ynol ether 6 ? and aryl alkyne 7
?,? readily afforded the ipso-coupling products 9aA and 10aA, respectively, each in 67% yield (FigureB,C).
Although the treatment of amides with an electrophilic activator leads to intermediates akin to those detected upon reaction of ynamides with an acid, ?,? diverging behavior is very often observed in their reactivity toward nucleophiles. ?,? We were pleased to find that amides such as 8 could nonetheless also be employed for this ipso-coupling process (FigureD), delivering α-branched aryl amide 11aA in good yield.
Aware of the ability of enantioenriched substrates to transfer their stereogenic information through sigmatropic rearrangements, ?,? we investigated the potential stereoselectivity of this transformation using both a chiral ynamide (in combination with a racemic sulfoxide) and a chiral sulfoxide (in combination with an achiral ynamide). Consistent with previous findings,? the use of a chiral ynamide did not lead to significant stereoselectivity, affording the product with a 1.5:1 dr (see SI, section 6.2, for details). Pleasingly, employing enantioenriched sulfoxides ((+)- or (−)-1a, both >99:1 er) in combination with 2D or 3A, the reaction proceeded with promising levels of chirality transfer, delivering (+)-4aD and (−)-5aA with 90:10 and 86:14 enantiomeric ratio, respectively (FigureA).
From a mechanistic standpoint, we initially proposed that vinyl cation derivative A, formed by protonation of an electron-rich alkyne (2, 3, 6 or 7), reacts with a sulfoxide nucleophile (1) to form oxasulfonium adduct B (FigureB). Given the results of FigureE, it appears that redistribution of electron density and polarization of the aryl moiety (represented by the resonance structure B′) is what allows circumvention of [3,3]-rearrangement reactivity. The representation as B′ highlights the ylidic nature of the C–S bond, foreshadowing the formation of C through a [2,3]-sigmatropic event.
To gain a deeper understanding of this rerouted sulfonium-rearrangement, computational studies were performed (RI-MP2-COSMO/def2-QZVP//RI-MP2/def2-SVP ?−? ? ? ? level of theory, see SI for the details). Importantly, considering our previous results,? we found it necessary to go beyond the standard density functional theory (DFT) calculations to the more advanced and less system-dependent wave function-based method (RI-MP2).
FigureC shows the computed Gibbs free energy profile starting from intermediate B (akin to an N,O-ketene acetal), which is formed by the attack of a sulfoxide on the transient keteniminium intermediate (A, where Y = N) in the initial stages of the reaction. ?,? Interestingly, in contrast to our initial proposal, the rearrangement was found not to proceed by a concerted mechanism.? Instead, cleavage of the S–O bond of intermediate B (via transition state TS _ B–I _) first leads to the formation of transient intermediate I, an initially unforeseen species (cf. FigureB), where the recently formed fragments are still closely associated. Even though many sulfonium rearrangements are believed to proceed through an asynchronous, concerted rearrangement in which S–O cleavage slightly precedes C–C bond formation, several examples exist in which both processes are separated by an intermediate. ?,?−? ? The rearrangement described herein seems to belong to the same category, a feature likely responsible for the regio-diverted outcome of the reaction. ?−? ? ?,?,?
Following the formation of intermediate I, a very low activation barrier (1.7 kcal mol^–1^) allows C–C bond formation, giving rise to intermediate C via TS _ I–C _. Early transition state TS _ I–C _ is characterized by the concurrent formation of a new C–C bond at the ipso-position and the breaking of weak interactions between the newly formed carbonyl and sulfide in transient intermediate I. Ultimately, ipso-substituted cationic intermediate C evolves into re-aromatized product D by S_N_2 attack of a thiol on the sulfide, with the simultaneous formation of a disulfide species (detected experimentally; see SI).
Furthermore, calculations strongly suggest that a direct [3,3]-sigmatropic rearrangement starting from this intermediate (B) is not possible, as no transition state leading to a potential intermediate E could be found (FigureC, see SI).
In conclusion, we have developed an acid-catalyzed strategy for the synthesis of α-arylated carbonyl compounds that allows buildup of sterically congested motifs. Our approach hinges on electron redistribution in an ipso-selective sulfonium rearrangement and is orthogonal to conventional transition metal-catalyzed cross-coupling logic. Computational studies revealed an unexpected intermediate in the proposed rearrangement process.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Johansson C. C. C.Colacot T. J.Metal-Catalyzed α-Arylation of Carbonyl and Related Molecules: Novel Trends in C–C Bond Formation by C–H Bond Functionalization Angew. Chem., Int. Ed.201049467670710.1002/anie.20090342420058282 · doi ↗ · pubmed ↗
- 2Hamann B. C.Hartwig J. F.Palladium-Catalyzed Direct α-Arylation of Ketones. Rate Acceleration by Sterically Hindered Chelating Ligands and Reductive Elimination from a Transition Metal Enolate Complex J. Am. Chem. Soc.199711950123821238310.1021/ja 9727880 · doi ↗
- 3Hamann B. C. H.Hartwig J. F.Palladium-Catalyzed Direct α-Arylation of Ketones. Rate Acceleration by Sterically Hindered Chelating Ligands and Reductive Elimination from a Transition Metal Enolate Complex J. Am. Chem. Soc.199711950123821238310.1021/ja 9727880 · doi ↗
- 4Shaughnessy K. H.Hamann B. C.Hartwig J. F.Palladium-Catalyzed Inter- and Intramolecular α-Arylation of Amides. Application of Intramolecular Amide Arylation to the Synthesis of Oxindoles J. Org. Chem.199863196546655310.1021/jo 980611 y · doi ↗
- 5Culkin D. A.Hartwig J. F.Palladium-Catalyzed α-Arylation of Carbonyl Compounds and Nitriles Acc. Chem. Res.200336423424510.1021/ar 020110612693921 · doi ↗ · pubmed ↗
- 6Hao Y.-J.Hu X.-S.Zhou Y.Zhou J.Yu J.-S.Catalytic Enantioselective α-Arylation of Carbonyl Enolates and Related Compounds ACS Catal.202010295599310.1021/acscatal.9b 04480 · doi ↗
- 7Ostrowska S.Scattolin T.Nolan S. P.N-Heterocyclic Carbene Complexes Enabling the α-Arylation of Carbonyl Compounds Chem. Commun.202157364354437510.1039/D 1CC 00913 C 33949497 · doi ↗ · pubmed ↗
- 8Lou S.Fu G. C.Nickel/Bis(Oxazoline)-Catalyzed Asymmetric Kumada Reactions of Alkyl Electrophiles: Cross-Couplings of Racemic α-Bromoketones J. Am. Chem. Soc.201013241264126610.1021/ja 909689 t 20050651 PMC 2814537 · doi ↗ · pubmed ↗