Familial novel androgen receptor gene variant associated with bilateral cryptorchidism and severe male infertility: A case report
Zohor Azher

TL;DR
A new androgen receptor gene variant is linked to cryptorchidism and infertility in two brothers, highlighting the importance of genetic testing.
Contribution
A novel AR gene variant (p.Tyr364His) is identified in two brothers with cryptorchidism and infertility, expanding the known mutational spectrum.
Findings
A novel AR missense variant (p.Tyr364His) was found in two brothers with cryptorchidism and infertility.
The variant is located in the N-terminal domain and is predicted to cause partial receptor dysfunction.
This case expands the AR mutational spectrum and underscores the need for early genetic evaluation in cryptorchidism.
Abstract
Cryptorchidism is a common congenital anomaly linked to infertility and testicular cancer risk. Variants in the androgen receptor (AR) gene cause androgen insensitivity syndrome (AIS), ranging from complete (CAIS) to partial (PAIS) and mild (MAIS) forms. We report a male patient with infertility, severe oligoasthenoteratozoospermia, and bilateral cryptorchidism. Whole-genome sequencing revealed a novel AR missense variant (p.Tyr364His) in the N-terminal domain, predicted to cause partial receptor dysfunction. The same variant was found in his brother with cryptorchidism and PAIS features. This finding expands the AR mutational spectrum and emphasizes the need for early genetic evaluation and counseling in cryptorchidism.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
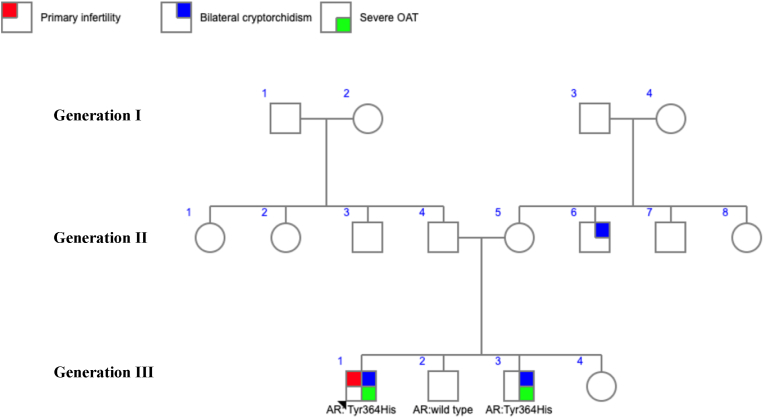 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSexual Differentiation and Disorders · Testicular diseases and treatments · Hormonal and reproductive studies
Introduction
1
Cryptorchidism, or undescended testis, refers to the failure of one (unilateral) or both testes (bilateral) to descend into the scrotal position. It is among the most common congenital anomalies in male newborns, with a reported prevalence of 3.4–5.8 % in full-term infants and up to 30 % in premature neonates.1 However, spontaneous testicular descent occurs in approximately 80 % of affected infants during the first three months of life, thereby reducing the true incidence to around 1 % by that age.2
Untreated or late-treated cryptorchidism patients have significant long-term consequences, including a markedly increased risk of testicular germ cell tumors and impaired fertility. Spermatogenesis is often compromised in testes that remain undescended,3 and approximately 10 % of infertile men report a history of cryptorchidism and orchidopexy.4 The incidence of azoospermia in unilateral cryptorchidism is estimated at 13 %, whereas it may exceed 90 % in untreated bilateral cases,5 making cryptorchidism the most common etiological factor of spermatogenic impairment in adulthood. Given the high incidence of cryptorchidism and its serious complications, routine neonatal screening through genital exam and early treatment is strongly recommended for cryptorchid boys.
From an embryological perspective, testicular descent occurs in two distinct phases. The transabdominal phase (weeks 8–15 of gestation) is primarily driven by insulin-like factor 3 (INSL3), secreted by fetal Leydig cells, which promotes transabdominal migration of the gubernaculum (caudal genital ligament). At the same time, androgens contribute indirectly by inducing regression of the craniosuspensory ligament, thus facilitating testicular descent. The subsequent inguinoscrotal phase (weeks 25 to term) is predominantly androgen-dependent, aided by additional factors such as intra-abdominal pressure leading to anchoring of fetal testes into the scrotum.6 Disruption of these pathways, whether due to abnormal secretion or action of INSL3 or androgen hormones, results in failure of normal testicular migration within the scrotum.
Here, we report the clinical presentation and genetic findings of two brothers with severe oligoasthenoteratozoospermia and bilateral cryptorchidism. Whole-genome sequencing (WGS) revealed a novel missense variant in the androgen receptor (AR) gene, providing further insight into the molecular mechanisms underlying this condition.
Case description
1.1
A 30-year-old man presented with a two-year history of primary infertility, associated with anorgasmia and erectile dysfunction since marriage. His past medical history was remarkable for bilateral cryptorchidism, managed by right orchiectomy and left orchidopexy at the age of three years. He denied any history of testicular trauma, mumps orchitis, radiation, or chemotherapy, and reported normal pubertal development.
Family history revealed non-consanguineous parents (Fig. 1). One brother (Individual III-2) had no history of cryptorchidism or infertility, while another brother (Individual III-3) had bilateral cryptorchidism corrected at age of three years; he is unmarried, thus his fertility status is undetermined.Fig. 1. Pedigree of the family with the AR (c.1090T > C; p. Tyr364His) variant. Arrow: the proband. Severe OAT: Severe oligoasthenoteratozoospermia. AR: Androgen receptor gene.Fig. 1
Physical examination of the proband showed normal body hair distribution and phallus, with the left testis palpable within a tight scrotum. No clinical evidence of congenital vasal agenesis was observed. Semen analyses repeatedly demonstrated markedly reduced sperm count, poor motility, and abnormal morphology, consistent with severe oligoasthenoteratozoospermia (OAT) (Table 1). Hormonal evaluation showed elevated FSH and LH with normal testosterone levels (Table 1). Scrotal ultrasound showed the left testicle is small in size, measuring 2.7 x 2.5 × 1 cm. Cytogenetic analysis revealed a male karyotype (46,XY) with no chromosomal abnormalities, and Y-chromosome microdeletion testing was negative. Two assisted reproductive technology (ART) attempts with intracytoplasmic sperm injection (ICSI) using ejaculated sperm were unsuccessful.Table 1. Semen analysis and hormone profile of a) the proband (Individual III-1) and b) his brother (Individual III-3). Semen analysis is performed on two separate occasions at least two months apart. FSH, follicle-stimulating hormone; LH, luteinizing hormone; T, total testosterone; PRL, prolactin.Table 1(a) Proband (Individual III-1):Semen Parameters1st Results2nd ResultsHormone profileResultsVolume (ml)0.52FSH (mIU/ml)43.40Sperm concentration (per ml)700,000500,000LH (mIU/ml)14.84Total motility (%)10 %11 motile sperm/slideT (nmol/ml)20.99Normal morphology (%)1 %0 %PRL (ng/ml)9.91(b) Brother (Individual III-3):Semen Parameters1st Results2nd ResultsHormone profileResultsVolume (ml)11FSH (mIU/ml)3.50Sperm concentration (per ml)800,0004,000,000LH (mIU/ml)5.51Total motility (%)40 %40 %T (nmol/ml)18.20Normal morphology (%)1 %1 %PRL (ng/ml)16.16
WGS was subsequently performed on DNA extracted from a buccal swab of the proband. A hemizygous missense variant in the AR gene (NM_000044.6: c.1090T > C; p.Tyr364His) was identified, predicted to substitute tyrosine with histidine at codon 364. This variant is rare, with a very low minor allele frequency (MAF) of 0.0000123 in gnomAD. Multiple in silico tools predicted a deleterious effect (Table 2).Table 2. In silico prediction tools and population databases of familial AR variant (p. Tyr364His). ACMG: American College of Medical Genetics guidelines.Table 2. Gene/Sequence IDAR/NM_000044.6DNA changec.1090T > CProtein changep. Tyr364HisSIFTDamagingPolyPhen-2Probably damagingCADD(PHRED)25.3REVEL0.67GenomAD0.0000123ACMG criteriaPM2, PP1, PP3, PP4
Segregation analysis revealed the AR variant in Individual III-3, while it was absent in Individual III-2. The maternal sample was unavailable for testing. A subsequent clinical evaluation of Individual III-3 revealed a normal general examination and phallus, with testicular volumes of 8 cc (right) and 18 cc (left). His semen analysis revealed abnormal parameters consistent with severe OAT. Hormonal evaluation was within normal limits, except for a mild elevation in prolactin (Table 1).
Discussion
2
AR gene, located on the long arm of the X chromosome (Xq12), encodes the androgen receptor protein, which is a member of the nuclear steroid receptor superfamily that functions as a transcriptional regulatory factor.7^,^8 Upon binding to steroid hormones (androgens) such as testosterone or dihydrotestosterone, the receptor undergoes activation and forms a hormone–receptor complex. This complex then dissociates from accessory proteins, translocates into the nucleus, where it regulates the transcription of androgen-responsive genes.9 These genes play a crucial role in male sex differentiation during embryogenesis (masculinization), the development of secondary sexual characteristics at puberty (virilization), and spermatogenesis.
Androgen insensitivity syndrome (AIS) is the most common disorder of sexual development (DSD) in individuals with a 46, XY karyotype.10^,^11 It is an X-linked recessive condition caused by pathogenic variants in the AR gene, which result in androgen receptor dysfunction and, consequently, resistance to androgen action. Based on clinical heterogeneity, AIS is classified into three phenotypes: complete androgen insensitivity syndrome (CAIS), partial androgen insensitivity syndrome (PAIS), and mild androgen insensitivity syndrome (MAIS). CAIS occurs in approximately 2–5 per 100,000 genetic males, while PAIS is thought to be at least as common. In contrast, MAIS is much less frequently reported.12 Individuals with CAIS typically present with normal female external genitalia with the absence of internal female genital structures and intra-abdominal, inguinal, or labial testes.13 PAIS is the incomplete form of androgen resistance and represents 10 % of individuals with AIS,14 which results from the partial inability of body cells to respond to androgens. The PAIS clinical manifestations vary according to the degree of AR residual function. Phenotypes range from predominantly female with evidence of external genital masculinization (such as clitoromegaly or posterior labial fusion) to predominantly male with genital anomalies including hypospadias, cryptorchidism, or micropenis, often associated with gynecomastia and impaired spermatogenesis. Patients with MAIS usually exhibit typical male external genitalia but may develop gynecomastia and infertility in adulthood.13 In the present report, the two brothers exhibited bilateral cryptorchidism and severe OAT, consistent with a partial phenotype of AIS.
The reproductive hormone profile in CAIS and PAIS patients is similar.15^,^16 It is characterized by elevated or normal serum testosterone levels associated with normal or high serum Luteinizing hormone (LH) levels reflecting androgen receptor dysfunction with normal androgen secretion.17 In AIS patients, follicle-stimulating hormone (FSH) and estradiol levels tend to be normal or slightly elevated for males.11 The proband in the present family demonstrated elevated FSH and LH levels with normal serum testosterone, a pattern consistent with both androgen resistance and impaired spermatogenesis. In contrast, the second affected sibling (III-3) showed a normal hormonal profile, which has also been reported in other AIS cases.
The AR gene spans approximately 90 kb of genomic DNA and comprises eight exons. It encodes the AR protein, a large polypeptide of 920 amino acids organized into four functional domains: the N-terminal transactivation domain (NTD), the DNA-binding domain (DBD), the hinge region (HR), and the ligand-binding domain (LBD). More than 1000 AR variants have been submitted in the AR database (ARDB; http://www.mcgill.ca/androgendb, updated September 2014), with approximately half reported in association with AIS and the remainder linked to other AR-related disorders, including spinal and bulbar muscular atrophy (Kennedy's disease), breast cancer, and premature ovarian failure18
AR variants are distributed across all exonic regions and affect each of the protein domains. The majority, however, are localized within the NTD (encoded by exon 1) and the LBD (encoded by exons 4–8). The NTD alone constitutes nearly half of the receptor and plays a critical role in regulating transcription of androgen-responsive genes. Consequently, structural or functional alterations in this domain significantly disrupt the androgen signaling pathway and often result in severe AR dysfunction. Indeed, most mutations in (exon1) have been associated with the CAIS phenotype. Nevertheless, distinct variants within the same coding region may also present with partial or mild forms of AIS.18 Frameshift and nonsense variants in the NTD introduce premature stop codons, leading to truncated, non-functional proteins and complete androgen receptor inactivation, thereby causing the CAIS phenotype.19 In contrast, missense variants in this domain may lead to either complete or partial androgen resistance, manifesting clinically as CAIS or PAIS, respectively.20 In the present family, we identified a novel missense variant, p.Tyr364His, within the NTD domain of the AR protein. This variant has not been previously reported and is predicted to partially impair AR function, thereby contributing to the PAIS phenotype. Furthermore, this variant expands the mutational spectrum of AR and further supports the prior evidence that missense variants in NTD can lead to an incomplete form of androgen resistance.
Conclusion
3
This study reports a novel familial variant in the AR gene, underscoring the importance of genetic evaluation in newborns presenting with cryptorchidism, with or without associated systemic manifestations. Early molecular diagnosis not only enables accurate prognosis but also facilitates the identification of related complications such as infertility. Furthermore, we emphasize the value of genetic screening in at-risk family members to allow for early diagnosis, appropriate counseling, and timely clinical management.
Author contributions
The author (corresponding author) was solely responsible for the conception and design of the study, collecting and interpreting clinical and genetic data, writing the manuscript draft, reviewing and editing the final version.
Funding sources
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kolon T.F.Patel R.P.Huff D.S.Cryptorchidism: diagnosis, treatment, and long-term prognosis Urol Clin 3132004 Aug 46948010.1016/j.ucl.2004.04.00915313056 · doi ↗ · pubmed ↗
- 2Khatwa U.A.Menon P.S.N.Management of undescended testis Indian J Pediatr 6762000 Jun 44945410.1007/BF 0285946610932966 · doi ↗ · pubmed ↗
- 3Foresta C.Zuccarello D.Garolla A.Ferlin A.Role of hormones, genes, and environment in human cryptorchidism Endocr Rev 2952008 Aug 156058010.1210/er.2007-004218436703 · doi ↗ · pubmed ↗
- 4Grasso M.Buonaguidi A.Lania C.Bergamaschi F.Castelli M.Rigatti P.Postpubertal cryptorchidism: review and evaluation of the fertility Eur Urol 202199112612810.1159/0004716801684324 · doi ↗ · pubmed ↗
- 5Lee P.A.Fertility in cryptorchidism. Does treatment make a difference?Endocrinol Metab Clin N Am 2231993 Sep 4794907902276 · pubmed ↗
- 6Rey R.A.Early orchiopexy to prevent germ cell loss during infancy in congenital cryptorchidism. Vol. 97J Clin Endocrinol Metab 20124358436110.1210/jc.2012-366223223482 · doi ↗ · pubmed ↗
- 7Pratt W.B.Welsh M.J.Chaperone functions of the heat shock proteins associated with steroid receptors Semin Cell Biol 521994 Apr 839310.1006/scel.1994.10127915146 · doi ↗ · pubmed ↗
- 8Quigley C.A.Bellis A.D.E.Marschke K.B.El-Awady M.K.Wilson E.M.French F.S.Androgen receptor defects: historical, clinical, and molecular perspectives Endocr Rev 1631995 Jun 27132110.1210/edrv-16-3-2717671849 · doi ↗ · pubmed ↗