Bone Marrow Failure Associated With Short Telomeres and Digenic Variants of Uncertain Significance in Telomere Biology Genes
Akhila Vadivelan, Geraldine Aubert, Julian A. Martinez, Alan Ikeda, Satiro De Oliveira, Theodore B. Moore, Vivian Chang

TL;DR
Two patients with bone marrow failure had short telomeres and inherited genetic variants in telomere-related genes, suggesting a possible digenic cause.
Contribution
The study highlights digenic inheritance of telomere biology disorders through variants of uncertain significance.
Findings
Two patients with bone marrow failure had very low telomere length and inherited VUS in multiple telomere genes.
Parents of the patients also showed low telomere length but no symptoms, indicating possible asymptomatic carrier status.
The findings suggest that synergistic VUS in different genes may lead to a penetrant disease phenotype.
Abstract
Telomere Biology Disorders (TBD) are a group of heritable disorders characterized by short telomeres. We report two patients that presented with bone marrow failure (BMF), who were identified to have low telomere length (TL) and variants of uncertain significance (VUS) in two different telomere genes inherited from their parents. At age 6, Patient 1 had stage 4 neuroblastoma. He was treated with chemotherapy, surgery, immunotherapy, and autologous stem cell rescue. At age 10, he developed pancytopenia. Bone marrow biopsy revealed hypocellular marrow and der (1; 7), associated with myelodysplastic syndrome. Germline genetic evaluation showed a pathogenic variant in DNAJC21 (from father) and VUS in NAF1 (c.1375C > T) (from father) and RTEL1 (c.533T > C) (from mother). The patient was found to have very low TL (VLTL) (< 1st percentile) in 4/6 white blood cell subsets. The patient's mother…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
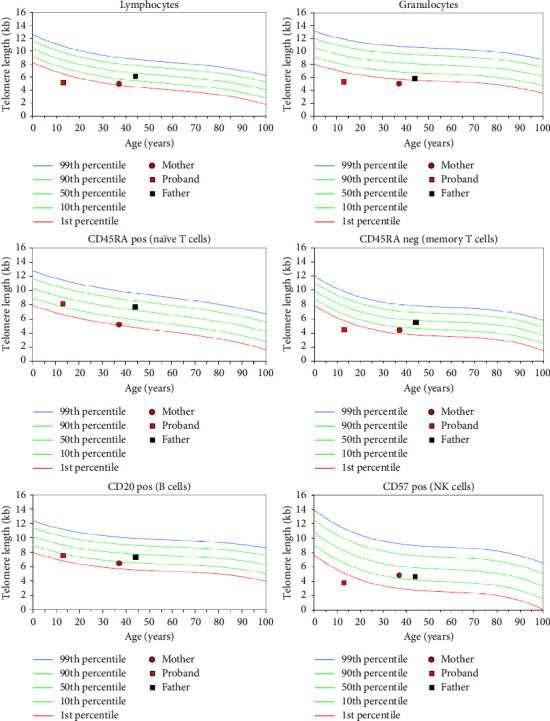 Figure 1
Figure 1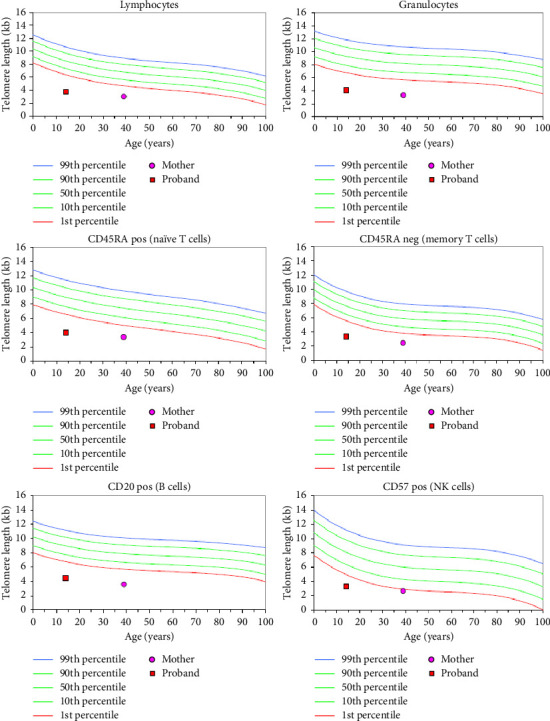 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTelomeres, Telomerase, and Senescence · Nuclear Structure and Function · Epigenetics and DNA Methylation
1. Introduction
Telomere Biology Disorders (TBD) are a heterogeneous group of heritable disorders characterized by abnormally short telomeres. Telomeres are DNA sequences present at the ends of the chromosomes that interact with shelterin complex proteins to protect the rest of the chromosome from appearing as a DNA break. When telomeres reach a certain length, cells stop dividing and become apoptotic or senescent. There are at least 17 genes associated with TBD including ACD, CTC1, DKC1, DCLRE1B, NHP2, NOP10, PARN, POT1, RPA1, RTEL1, STN1, TERC, TERT, TINF2, WRAP53, NAF1, and ZCCHC8. These genes encode components of telomerase or encode proteins that are involved in telomerase assembly, transportation to the telomere, or docking with the telomere end, that when mutated, result in telomeres being too short. These genes are inherited in an autosomal dominant, autosomal recessive, or X-linked recessive pattern, and several genes have been reported to be autosomal dominant and autosomal recessive [1].
Patients with TBD present with a wide variety of features affecting nearly every organ and system. They are at increased risk of myelodysplastic syndrome (MDS), bone marrow failure (BMF), leukemia, cancers of the head, neck and genitourinary system, as well as pulmonary fibrosis, emphysema, and liver fibrosis/cirrhosis [1–4]. Dyskeratosis congenita (DC) is a classic presentation of TBD during childhood where patients have a triad of abnormal skin pigmentation, nail dystrophy, and oral leukoplakia [5]. However, one review determined that less than half of patients with TBD presented with all three classic features of DC. Patients with Revesz Syndrome (RS) also present in early childhood with DC features and bilateral exudative retinopathy [6]. Hoyeraal–Hreidarsson Syndrome (HH) is defined as symptoms of DC and cerebellar hypoplasia [7]. Coats plus syndrome is another form of TBD characterized by intracranial calcifications, leukodystrophy, and brain cysts [8]. Additionally, TBDs can initially present in adulthood as isolated pulmonary, liver, or hematologic disorders, without prior childhood manifestation. These patients often have telomere lengths (TLs) in the 1^st^–10^th^ percentile ranges and without identifiable germline genetic changes [9]. The spectrum of TBD phenotypes and the challenges of diagnosing TBDs suggest that genotype-phenotype correlations are complex. In fact, a recent publication identified polygenic modifiers of TL in patients with monogenic TBD-associated mutations [10].
TBD can be expressed in various ways, genetic anticipation has been described in some families with more severe symptoms appearing in each successive generation. Phenocopying has also been observed in families with TBD, where the phenotype is dissociated from the genotype. In such individuals, despite inheriting a wild type gene from their parents, they show symptoms of TBD from short telomeres. This implies that an absence of mutation does not mean absence of risk [11]. Variable expressivity and incomplete penetrance of TBD has been reported especially in families with pulmonary disease [12, 13]. Polygenic modifiers have also been noted in TBD [14].
We report on two patients with clinical manifestations of TBD, with short telomeres and variants of uncertain significance (VUS) in two different telomere genes inherited from their parents. These cases highlight the importance of TL testing and suggest complex genetic factors leading to a TBD diagnosis, such as digenic inheritance, genetic anticipation, unknown genetic modifiers, and potential environmental factors.
2. Methods
We performed an observational study of two patient families that presented to our clinic. We interviewed the proband and their families. We performed clinical genetic testing as detailed below in the respective cases. We then analyzed the genetic results, correlated with the clinical presentation. No written consent has been obtained from the patients as there is no patient identifiable data included in this case report/series.
2.1. Case 1
Patient 1 was initially diagnosed with stage 4 neuroblastoma at the age of six. He was treated with chemotherapy that included cyclophosphamide, topotecan, and etoposide, as well as surgery, immunotherapy, and autologous stem cell rescue. The patient completed therapy at the age of nine with complete remission. During his cancer survivorship care, he was diagnosed with genu varum.
At the age of 10 years, the patient developed frequent and severe epistaxis. Upon evaluation, he was found to have thrombocytopenia and anemia. He was treated with platelet, red blood cell transfusions, and with intravenous immunoglobulin (IVIG). Subsequently, he developed neutropenia. He underwent a bone marrow aspirate and biopsy which revealed der (1; 7) which is associated with myelodysplastic syndrome (MDS) [15], no dysplasia or recurrence of neuroblastoma and 50% cellular marrow with trilineage hematopoiesis. As the patient was symptomatically stable and this diagnosis was made during the COVID-19 pandemic, bone marrow transplant was deferred.
The patient underwent a 574-gene panel for “Inborn Errors of Immunity and Cytopenias” and was found to have one pathogenic variant in DNAJC21 and variants of uncertain significance (VUS) in NAF1 (c.1375C > T) and RTEL1 (c.533T > C) (Table 1). Cascade testing revealed that the DNAJC21 and NAF1 variants were inherited from father and RTEL1 variant was inherited from mother. Because NAF1 and RTEL1 are related to autosomal dominant or autosomal recessive TBD [1, 16], TL testing was performed. He was found to have very low TL (VLTL) < 1st percentile in the granulocytes, lymphocytes, memory T cells and NK cells (Figure 1). The patient's 40-year-old mother was found to have low TL 1 and < 10^th^ percentiles in her lymphocytes, naïve T cells, memory T cells, and B cells, and VLTL in her granulocytes. Other than migraines, she reported no health problems. Father was 46-year-old and had low TL 1 and < 10^th^ percentiles in granulocytes but normal TL in all other subsets. He had no known health problems. We interpreted these findings as consistent with the patient having TBD and the patient's mother either also having TBD or being a carrier, and the potential that genetic anticipation may be involved.
During the pretransplant evaluation of the patient, he was found to have a dilated aortic root measuring a Z score of +4.6. He then underwent reduced intensity conditioning using busulfan, fludarabine, and anti-thymocyte globulin for a 4/6 matched cord stem cell transplant. He engrafted within 10 days of the transplant and remained admitted for managing electrolyte imbalances and adjusting tacrolimus to therapeutic levels. He was discharged home on Day + 63. He was then re-admitted for evaluation of cervical lymphadenopathy on day +95. Biopsy results of cervical lymph node were inconclusive for infection or monotypic B cells, but pain and swelling decreased with antibiotics. He also developed hemolytic anemia secondary to adenovirus infection which was treated with steroids and rituximab. He is now off steroids and continues to be on low-dose tacrolimus on day +260.
2.2. Case 2
Patient 2 is a 15-year-old male with acne vulgaris who presented with a depressed skull fracture of the parietal bone and scalp hematoma following a football head injury. He was incidentally found to have a platelet count of 69 × 10^3^/μL, which delayed neurosurgical intervention. Initially, his thrombocytopenia was attributed to immune thrombocytopenic purpura. However, IVIg was given twice with no significant response. He was also noted to have mild anemia with macrocytosis and mild leukopenia. He had no history of easy bruising or bleeding and tolerated an appendectomy without excessive bleeding. Family history was notable for a paternal uncle with chronic myeloid leukemia in his 30s.
Bone marrow biopsy revealed a hypocellular marrow with multilineage maturation, mild fibrosis, and no genetic abnormalities. Peripheral blood flow cytometry was negative for any blasts. He was then admitted to our hospital and transfused 3 units of platelets which resulted in an increase in platelet count to > 100 × 10^3^ μL to undergo craniotomy for cranial vault reconstruction. He tolerated the procedure well.
During workup for aplastic anemia, the patient was tested for TL which was shortened below 1^st^ percentile in all 6 white blood cell subsets (Figure 2). He underwent clinical trio exome sequencing along with his parents. He was found to have three VUSs in TERT (c.3158-80G > A) (inherited from mother), TINF2 (c.814T > C) (inherited from mother) and SRP72 (c.1928C > T) (inherited from father) (Table 1). Mother was 42-year-old and found to have VLTL (< 1^st^ percentile) in all 6 white blood cell subsets. She reported early greying in her 30 s, hyperthyroidism, and poor dentition. Her complete blood counts, liver numbers, and pulmonary function tests have remained normal. Father was 43-year-old and had no known health problems.
Patient continues to be followed up in our cancer predisposition clinic and continues to have pancytopenia with latest Hb at 13.9 g/dL, WBC at 3.09 × 10^3^ μL and platelets at 52 × 10^3^ μL but does not require any transfusion support at this time.
3. Discussion
In the first case, the patient was diagnosed with MDS at the age of 14 and found to have short telomeres and VUSs in RTEL1 and NAF1. RTEL1 has been shown to be autosomal recessive or dominant, while NAF1 has been shown to be autosomal dominant [1]. The patient's RTEL1 variant was inherited from mother, who has VLTL in the granulocyte compartment and low TLs in lymphocytes, native T cells, memory T cells, and B cells, which could be consistent with the patient's mother either having TBD or being a carrier of TBD, with possible anticipation occurring in our patient who has history of cancer, MDS, and VLTL [1]. The NAF1 variant was inherited from father who displayed normal TLs, except in granulocytes where his TL ranked between 1^st^ and 10^th^ percentiles.
Interestingly, this patient also had a heterozygous pathogenic variant in DNAJC21, which encodes a heat shock protein that may play a role in ribosomal RNA biogenesis, inherited from father [17]. Biallelic germline variants in DNAJC21 cause Schwachman Diamond Syndrome (SDS), which is characterized by short stature, exocrine pancreatic dysfunction, skeletal abnormalities, and BMF [18]. Although carriers of SDS are thought to be asymptomatic, our patient did have genu varum, a skeletal defect of the knees that has been reported in SDS [18, 19]. Genu varum is a rare diagnosis after the age of 3-4 years and can be associated with rickets or skeletal disorders [20–22]. Patients with SDS were shown to have relatively short TL, which raises the possibility of inheritance of short TL from the carrier parent that could contribute to genetic anticipation [23]. It is possible that this patient's DNAJC2 carrier status could be synergistic with the VUSs in NAF1 and RTEL1. The chemotherapy that this patient received for neuroblastoma may also have contributed to the development of the MDS, in addition to an underlying TBD.
Similarly, in the second case, the patient developed abnormal blood counts at the age of 15 years and was found to have short telomeres and VUSs in TERT and TINF2, both inherited from mother, who shares VLTLs in all 6 measured subsets. Both TERT and TINF2 are inherited in an autosomal dominant manner. TINF2 mutations were first described in a family with genetic anticipation [24] but germline variants in TINF2 are almost always de novo and not inherited as they are in this patient [1]. This suggests that this patient's variant in TINF2 could be presenting as a milder form of TBD but, in conjunction with TERT, playing a synergistic effect. All patients described with TINF2 mutations are located on exon 6 such as our patient. Alternatively, it is possible that the TERT VUS is truly pathogenic on its own.
This patient also inherited a VUS in SRP72 from their father. SRP72 encodes signal recognition particle 72, which mediates the targeting of secretory proteins to the endoplasmic reticulum [25, 26]. Heterozygous germline variants in SRP72 have been associated with familial myelodysplasia [27], but the patient's father has no signs or symptoms of any blood abnormalities.
Identifying underlying TBD in patients who present with BMF is critical for clinical management. Allogeneic stem cell transplant is curative for several hematologic conditions that complicate TBD but requires careful donor selection, modified conditioning regimen, and special consideration of the many organ toxicities that may occur [28, 29]. Additionally, patients with TBD require life-long surveillance for cancer and assessments for optimal immune function, nutrition, bone health, lung function, liver disease, ophthalmologic health, and mental health [30].
These two patient families each have more than one variant of uncertain significance in at least two telomere-related genes, highlighting the complexities of underlying causes of TBD and short TL. These cases suggest possible digenic inheritance, which has been described in several recessive conditions, including familial hemophagocytic lymphohistiocytosis (HLH) [31], retinitis pigmentosa [32], holoprosencephaly [33], and others [34]. Synergy can occur in genetic conditions where two variants contribute to a phenotype and can result in earlier and severe onset of disease [35]. Often synergy occurs when these variants involve genes with functional overlap or genes involved in the same pathway, as seen in HLH, a disorder typically caused by biallelic variants in genes associated with the function of cytotoxic lymphocytes [36]. In one review of over 2700 patients suspected to have familial HLH, there were 21 patients with heterozygous variants in PRF1 and a degranulation gene, and 7 patients with heterozygous variants in 2 different genes involved in degranulation [37]. Another contributing factor could be genetic anticipation.
4. Conclusion
Our two cases with germline variants in different genes regulating TL suggest a digenic mode of inheritance in TBD. Larger cohort studies are needed to determine the prevalence of this mode of inheritance.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Revy P. Kannengiesser C. Bertuch A. A. Genetics of Human Telomere Biology Disorders Nature Reviews Genetics 20232428610810.1038/s 41576-022-00527-z 36151328 · doi ↗ · pubmed ↗
- 2Savage S. A. Bertuch A. A. The Genetics and Clinical Manifestations of Telomere Biology Disorders Genetics in Medicine 2010121275376410.1097/gim.0b 013e 3181 f 415b 52-s 2.0-7865063912621189492 PMC 3825100 · doi ↗ · pubmed ↗
- 3Yamada S. Misawa K. Mima M. Telomere Shortening in Head and Neck Cancer: Association Between DNA Demethylation and Survival Journal of Cancer 20211282165217210.7150/jca.5476033758594 PMC 7974875 · doi ↗ · pubmed ↗
- 4Renoux M. C. Mazars N. Tichit R. Counil F. Cyanosis Revealing Hepatopulmonary Syndrome in a Child With Dyskeratosis Congenita Pediatric Pulmonology 20104519910210.1002/ppul.211282-s 2.0-7424912247819953657 · doi ↗ · pubmed ↗
- 5Savage S. A. Alter B. P. Dyskeratosis Congenita Hematology-Oncology Clinics of North America 200923221523110.1016/j.hoc.2009.01.0032-s 2.0-6264910721419327580 PMC 2702847 · doi ↗ · pubmed ↗
- 6Karremann M. Neumaier-Probst E. Schlichtenbrede F. Revesz Syndrome Revisited Orphanet Journal of Rare Diseases 2020151 p. 29910.1186/s 13023-020-01553-y PMC 758328733097095 · doi ↗ · pubmed ↗
- 7Glousker G. Touzot F. Revy P. Tzfati Y. Savage S. A. Unraveling the Pathogenesis of Hoyeraal–Hreidarsson Syndrome, a Complex Telomere Biology Disorder British Journal of Haematology 2015170445747110.1111/bjh.134422-s 2.0-8493813937425940403 PMC 4526362 · doi ↗ · pubmed ↗
- 8Simon A. J. Lev A. Zhang Y. Mutations in STN 1 Cause Coats plus Syndrome and Are Associated With Genomic and Telomere Defects Journal of Experimental Medicine 201621381429144010.1084/jem.201516182-s 2.0-8498298570627432940 PMC 4986528 · doi ↗ · pubmed ↗