Curated mitochondrial genome reference database of state key protected wild mammal in China
Xia Huang, Guihua Zhang, Joseph D. Orkin, Shiyun Liu, Shan Jiang, Yinhui Zhao, Pengfei Fan, Lianghua Huang, Xiaoming Zhang, Xueyou Li, Song Li, Kai He, James Crainey, James Crainey, James Crainey

TL;DR
This paper creates a detailed mitochondrial genome database for protected wild mammals in China, improving species identification and conservation efforts.
Contribution
The study generates new mitochondrial genomes and updates the protected species list, addressing taxonomic and genetic data gaps.
Findings
A refined list of 169 protected mammalian species was established using updated taxonomic and distributional evidence.
12 new mitochondrial genomes were generated for nine taxa, filling critical genetic data gaps.
Taxonomic ambiguities and mislabeling issues were identified in genera like Moschus and Naemorhedus.
Abstract
Effective conservation of wild mammals necessitates accurate taxonomic classification and reliable genetic reference data. In China, the List of State Key Protected Wild Animals serves as a critical tool for species protection. However, taxonomic revisions and gaps in genetic data can impede its effectiveness. In this study, we updated the List of State Key Protected Wild Animals (2021) by incorporating recent taxonomic and distributional evidence, resulting in a refined list of 169 mammalian species that are protected. We identified 15 taxa lacking complete mitochondrial genome data and addressed this gap by generating 12 new mitogenomes for nine taxa using a combination of GenBank database mining and next-generation sequencing of museum specimens and fecal samples. These efforts led to the establishment of a curated mitochondrial genome reference database encompassing 164 species. Our…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
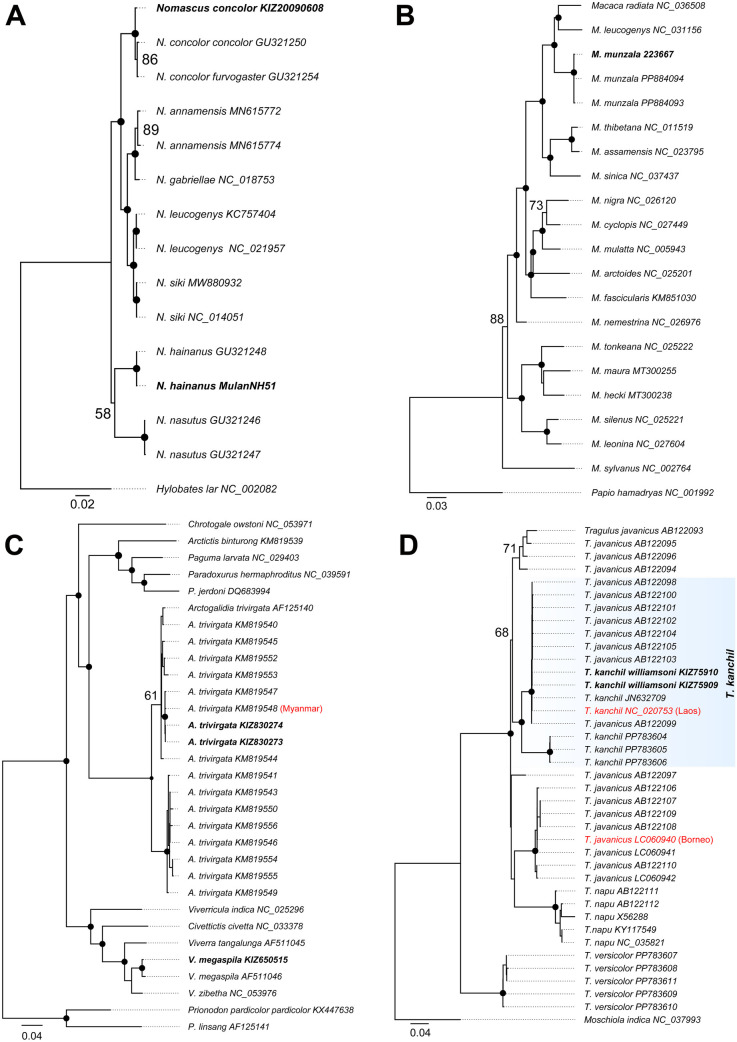 Fig 1
Fig 1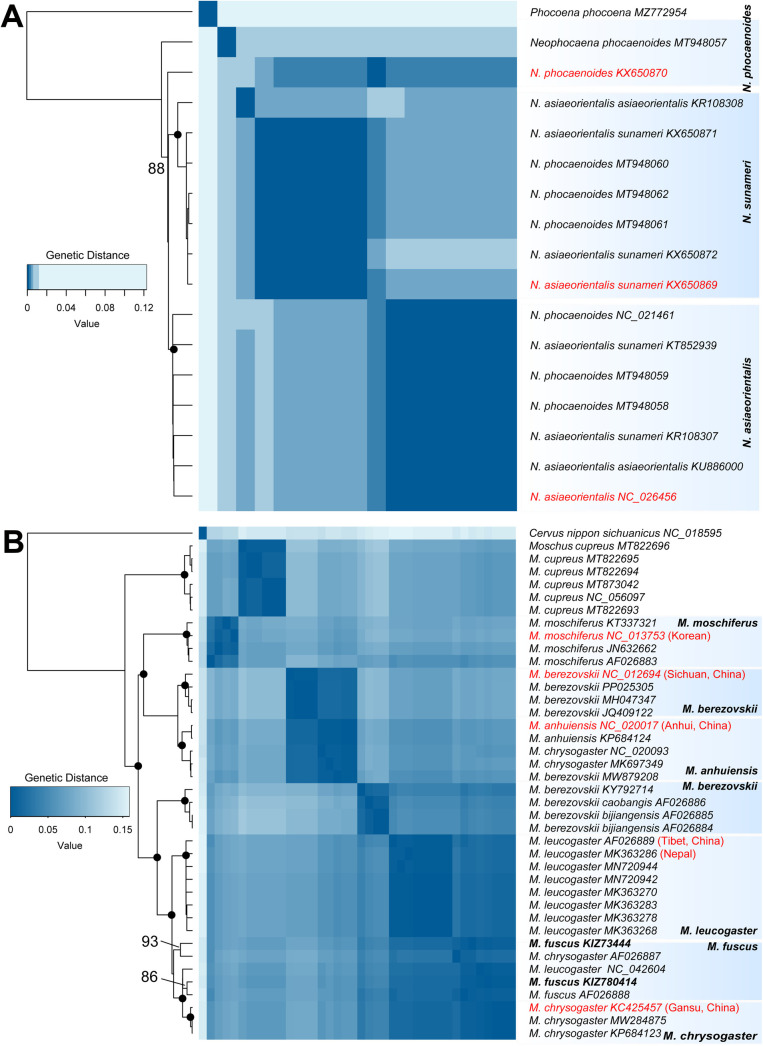 Fig 2
Fig 2- —http://dx.doi.org/10.13039/501100001809National Natural Science Foundation of China
- —http://dx.doi.org/10.13039/501100001809National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIdentification and Quantification in Food · Environmental DNA in Biodiversity Studies · Genomics and Phylogenetic Studies
Introduction
Wild mammals are integral to ecosystems, contributing to processes such as nutrient cycling, seed dispersal, pollination, and pest control [1]. However, climate change, habitat loss, fragmentation, and degradation, have led to significant population declines, pushing many species toward extinction [2–4]. In response to the accelerating biodiversity crisis, governments and conservation organizations have intensified efforts to protect vulnerable species, notably through the development and regular updating of species protection lists such as the International Union for Conservation of Nature (IUCN) Red List. To capture nation-specific conservation needs, the Chinese government has developed its national classification system: The List of State Key Protected Wild Animals (LSKPWA), which is updated approximately every five years. This list divides species into Class I and Class II based on their conservation urgency, legal status, and ecological importance, with Class I encompassing species at greater risk. The latest revision, published in 2021, expanded the list to include 980 species and eight categories, with 234 species and one category under Class I, and 746 species and seven categories under Class II (http://www.forestry.gov.cn/main/5461/20210205/122418860831352.html). In China, the legal penalties for the illegal hunting, killing, acquisition, transportation, or sale of wildlife vary depending on the species’ protection status.
Despite efforts to update LSKPWA, continuous taxonomic revisions have led to discrepancies between the LSKPWA and current species classifications in China. For instance, mammalian taxonomy has undergone extensive changes in recent years [5]. These revisions include the elevation of subspecies to full species [6], confirmation of certain species’ absence from China, and synonymization of species with existing taxa [7]. These taxonomic updates result in misalignments between species inventories and LSKPWA. For example, red pandas (Ailurus fulgens), which are listed as Class II in the 2021 LSKPWA, have been reclassified into two species: the Chinese red panda (A. styani) with a stable, widespread population and the Himalayan red panda (A. fulgens) with a restricted range and critically low population size [6]. Such misalignments can obscure conservation priorities and hinder effective law enforcement.
In addition to the misalignments, accurate species identification is further complicated by morphological similarities among certain species, despite differences in their conservation status. For instance, three species within the genus Martes are morphologically similar, but only one is listed under Class I, while the others fall under Class II. Distinguishing these species based solely on appearance is challenging, particularly for non-specialists or when dealing with incomplete specimens. Furthermore, confiscated wildlife materials often consist of partial remains (e.g., hides, bones, or frozen tissues) or processed products (e.g., musk, pangolin scales, ivory, or horn products), which lack diagnostic morphological features, necessitating molecular identification techniques [8,9].
DNA barcoding is a widely adopted tool for species identification in both ecological and forensic contexts [10,11]. Traditional barcoding typically uses short mitochondrial gene fragments, such as two ribosomal RNAs (12S rRNA and 16S rRNA), cytochrome c oxidase subunit 1 (COI), or cytochrome b (CYTB) [12,13]. While sufficient in most cases, these single-gene markers could be insufficient when closely related species exhibit low interspecific divergence. For instance, COI could not reliably distinguish between Procapra przewalskii and P. gutturosa, which exhibit only 0.5% genetic divergence in this gene [14]. In contrast, complete mitochondrial genome sequences provided clear phylogenetic resolution between the two species allowing for accurate identification [15]. This illustrates the need for complete mitochondrial genomes (mitogenomes) as a more comprehensive reference data.
Although GenBank hosts a growing collection of mitogenome sequences, gaps and inconsistencies remain. Our preliminary review revealed that at least 15 mammalian species or subspecies listed in China’s LSKPWA still lack complete mitogenomes, including multiple primates and ungulates (S1 Table). Moreover, public databases commonly include misidentification or mislabeling [16,17], which, if used uncritically, can lead to erroneous species identifications and legal misinterpretations.
To address these challenges, this study aims to establish a curated mitochondrial genome reference database for the state key protected wild mammals in China. The objectives include updating the list of protected species to incorporate current taxonomic and distributional data, identifying gaps in existing mitogenome datasets, and using next-generation sequencing technologies to generate new sequences. This curated database will serve as a crucial tool for forensic investigations, enhancing the accuracy of species identification in cases of wildlife trafficking, and other legal matters related to conservation.
Materials and methods
Updating the list of state key protected wild mammals in China
To update the taxonomy of species in the 2021 LSKPWA, we integrate the latest expert consensus from key national publications [5,7] and the internationally recognized Mammal Diversity Database v2.3 (https://zenodo.org/records/17033774; last update September 2, 2025), which is actively curated by the American Society of Mammalogists. Each revision was cross-referenced with the latest peer-reviewed publications and verified by relevant experts.
Taxon sampling, mitogenome sequencing and assembly
We screened the updated species list against GenBank and identified ten species and five subspecies lacking mitogenomes (S1 Table). These included four primates, two carnivores, one lagomorph, and eight cetartiodactylans.
We collected 10 samples representing seven species, including feces, museum skin, and tissue samples (Table 1). Genomic DNA from museum skins and tissue samples was extracted using the DNeasy Blood & Tissue Kit (Qiagen, Germany, Cat. No. 69504), while DNA from fecal samples was extracted with the QIAamp® Fast DNA Stool Mini Kit (Qiagen, Canada, Cat. No. 51604). Where necessary, genomic DNA was fragmented using NEBNext® dsDNA Fragmentase (New England Biolabs, Cat. No. M0348S). Double-stranded libraries were prepared using the VAHTS Universal DNA Library Prep Kit for Illumina V4 (Vazyme, Cat. No. ND610). DNA libraries generated using museum skins and tissue samples were directly sequenced on the Illumina HiSeq X platform, producing 2 × 150-bp reads. For fecal-derived DNA libraries, we employed a capture hybridization approach prior to sequencing [18]. Biotin-labeled probes targeting the mitogenome were prepared via long-range PCR amplification of the human mitochondrial genome [19], purified with the Zymoclean™ Gel DNA Recovery Kit (Zymo; Cat. No. D4007) and biotinylated using the Biotin-Nick Translation mix (Roche; Cat. No. 11745824910). Libraries were incubated with the probes and enriched mitochondrial DNA was sequenced as described above.
Table 1: Species sequenced in this study.
Quality control and trimming of all raw data were conducted using fastp with the parameters -g -q 20 -u 50 -n 15 [20]. Mitogenomes were initially assembled and annotated using Mitoz v3.6 with default parameters [21]. For two samples of species Moschus fuscus (KIZ780414) and Viverra megaspila (KIZ650515) with relatively lower coverage of the mitogenomes, and the genomic Pacbio sequencing data of Muntiacus vaginalis nigripes we also aligned the reads to an assembled mitogenome using BWA-MEM [22] and annotated using Geneious Prime® 2024.0.5 to cross verify the result. The raw genome-resequencing reads of Budorcas taxicolor white (SRX15185605) and Muntiacus vaginalis nigripes (SRR28810463) were downloaded from NCBI Sequence Read Archive (SRA) and processed similarly. All newly sequenced mitogenomes were verified manually and deposited in GenBank (Accession Numbers: PQ740948-PQ740958, BK070181).
Curation of mammalian mitogenome reference sequences
To develop a curated mitochondrial genome dataset, we retrieved available mitogenome sequences from GenBank. Given the potential for misidentified submissions in GenBank, we employed the phylogenetic species concept (PSC) [23] to delineate species. Specifically, we evaluated whether sequences attributed to a given species formed a monophyletic clade. The most reliable sequence for each species was designated as its reference sequence.
To facilitate this analysis, we downloaded all available mitochondrial CYTB and COI gene sequences from GenBank for each order, as these genes are the most extensively represented in mammalian studies. All sequences were aligned with their corresponding mitogenomes using MAFFT v7.490 [24]. Maximum-likelihood (ML) phylogenetic trees were then constructed using IQ-TREE v1.6.8 [25] within PhyloSuite v1.2.2 [26]. For phylogenetic analyses, tRNA genes, the NADH dehydrogenase subunit 6 (ND6) gene, and the control region (D-loop) were excluded.
In most cases, each mitogenome clustered with its conspecific sequences with high bootstrap support (BS) (i.e., BS ≥ 80), validating their taxonomic assignments. However, instances of paraphyly and polyphyly were observed in genera such as Naemorhedus and Moschus. For these cases, we examined sample localities and prioritized sequences from type localities as reference sequences. Pairwise genetic distances were calculated using the Kimura 2-parameter (K80) model, and genetic distance heatmaps were visualized using the R packages ape [27], seqinr [28], phangorn [29], and gplots (https://github.com/talgalili/gplots). Through this process, we established reliable reference mitogenome sequences for 164 species, ensuring accurate representation in our curated dataset.
Results
The updated list of state key protected wild mammals in China
The 2021 LSKPWA in China included 185 mammalian taxa. Following a comprehensive taxonomic revision and evaluation of species distribution, we determined that the updated list should include 169 valid species (S1 Table). This revision involved the removal of eight taxa that are either confirmed to be absent from China’s territory or have been extirpated in the wild within China. In addition, 11 taxa previously recognized as distinct species were reclassified as subspecies under known species. These include Ovis ammon (now including collium, darwini, hodgsoni, karelini, and polii as subspecies), Cervus elaphus (including wallichii and yarkandensis as subspecies), Muntiacus vaginalis (including nigripes), Tragulus kanchil (including williamsoni), Budorcas tibetana (including bedfordi) and Budorcas taxicolor (including whitei). Three taxa were synonymized with recognized species based on recent taxonomic evidence: Delphinus capensis was merged with Delphinus delphis; Cervus canadensis with Cervus elaphus; and Naemorhedus griseus with Naemorhedus goral [30,31]. Conversely, two former subspecies were elevated to full species status based on molecular and morphological studies: A. styani, previously considered a subspecies of A. fulgens [6] and Trachypithecus melamera, formerly treated as a subspecies of Trachypithecus phayrei (T. phayrei shanicus) [32]. In addition, we incorporated Mesoplodon hotaula (Deraniyagala’s Beaked Whale) into the updated list following its first confirmed record in the South China Sea [33]. Finally, the pygmy slow loris Nycticebus pygmaeus has been reassigned to the newly established genus and species Xanthonycticebus intermedius [34,35].
Phylogenetic relationships and species diversity
We successfully generated 12 new complete mitogenome sequences for eight species or subspecies either by using newly sequenced data (n = 10) or by extraction from the raw genome resequencing SRA reads (n = 2) (Table 1). Phylogenetic analyses of these new mitogenomes clustered them with their congeneric species validating their taxonomic classifications (Figs 1 and 2; S1 Fig). For example, the Hainan gibbon (Nomascus hainanus) and the Cao vit gibbon (N. nasutus) form sister taxa, occupying a basal position within the genus Nomascus (Fig 1A), being consistent with previous studies using CYTB gene [36]. The Arunachal macaque (Macaca munzala) was identified as the sister species to the M. radiata and M. leucogenys (BS = 100) (Fig 1B), corroborating earlier phylogenetic results based on D-loop and CYTB sequences [37,38]. The mitogenome of Large-spotted civet (Viverra megaspila; KIZ650515) formed a sister relationship with V. zibetha and V. tangalunga (Fig 1C), aligning with previous findings [39]. The two newly sequenced individual of the small-toothed palm civet (Arctogalidia trivirgata) clustered with a lineage previously identified in Myanmar (BS = 95), corresponding to the subspecies A. trivirgata millsi (Fig 1C). Two mitogenomes of T. kanchil williamsoni from Yunnan, China clustered within the clade of T. kanchil from Laos, affirming that T. williamsoni in China belongs to T. kanchil (Fig 1D). Similarly, the mitogenome of Budorcas taxicolor whitei clustered with other sequences of B. taxicolor, forming a well-supported sister lineage to the nominal subspecies B. t. taxicolor (BS = 100) (S1A Fig), being consistent with genomic evidence [40]. Finally, the mitogenome of Muntiacus vaginalis nigripes (SRR28810463), endemic to Hainan Island, clustered within M. vaginalis (BS = 100) (S1B Fig), supporting its treatment as a subspecies rather than a distinct species [41].
The Maximum-likelihood (ML) phylogenetic tree of species newly sequenced in this study based on mitochondrial genome dataset.(A) Nomascus concolor and N. hainanus; (B) Macaca munzala; (C) Arctogalidia trivirgata and Viverra megaspila. (D) Tragulus kanchil williamsoni. Nodes with BS ≥ 95 are indicated by black dots, while those with BS below 95 are shown as numbers. Bold names are newly obtained sequences in this study. Red names are designated curated mitochondrial genome sequence for species based on type locality and phylogenetic relationships. Sequences from GenBank used in this study are named with their accession numbers.
Genetic distance heatmaps based on the CYTB gene using the Kimura 2-parameter (K80) model, integrated with ML phylogenetic trees constructed from the mitochondrial genome dataset.(A) For the genus Naemorhedus, pairwise genetic distances between species range from 0.5% to 12.2%; (B) For the genus Moschus, pairwise genetic distances between species range from 0.9% to 15.6%. Nodes with BS ≥ 95 are indicated by black dots, while those with BS below 95 are shown as numbers. Bold names are newly obtained sequences in this study. Red names are designated curated mitochondrial genome sequences for a species based on type locality and phylogenetic relationships. Sequences from GenBank used in this study are named with their accession numbers.
We observed paraphyly and polyphyly in finless porpoises (Neophocaena) and musk deer (Moschus). Certain clades of Neophocaena encompassed sequences labeled as different species (Fig 2A), indicating misidentification or the use of out-of-date taxonomy. Despite this, our phylogenetic tree aligns well with a previous study [42], suggesting an updated classification recognizing three distinct species: the Yangtze finless porpoise (N. asiaeorientalis), the East Asian finless porpoise (N. sunameri), and the Indo-Pacific finless porpoise (N. phocaenoides).
Within the genus Moschus, our phylogenetic analyses revealed several instances of taxonomic ambiguity (Fig 2B). One clade grouped the topotype of M. anhuiensis with sequences labeled as M. berezovskii and M. chrysogaster (BS = 100), exhibiting low genetic divergence (< 1.0%), suggesting potential misidentification or historical gene flow. M. fuscus appeared in two distinct clades: one included a sample from Gongshan (KIZ73444) (BS = 93), and the other comprised a sample from Biluoxueshan (KIZ780414), a sequence labeled as M. leucogaster from Tibet (NC_042604), and M. chrysogaster from various localities (BS = 97). Notably, NC_042604 did not cluster with the topotype of M. leucogaster from Nepal, indicating possible misidentification. Additionally, samples of M. berezovskii formed two genetically distinct clades with a divergence of 5.9%: one from its type locality in Sichuan and another comprising subspecies bijiangensis and caobangis from Yunnan, suggesting unrecognized species-level diversity.
A curated mitogenome database of state key protected wild mammals in China
Based on phylogenetic analyses, assessments of monophyly and type localities, we designated reference mitogenomes for 164 species. Mitogenomes were unavailable for five species including M. peruvianus, M. hotaula, M. leucogaster, N. nasutus, and Ochotona iliensis, for which only partial yet reliable CYTB and/or D-loop sequences are available (S1 Table).
Discussion
This study enhances the taxonomic framework and mitogenomic resources for China’s state key protected wild mammals. We revised the 2021 LSKPWA to reflect recent taxonomic updates, resulting in a refined list of 169 mammalian species that are protected. By integrating publicly available data with 12 newly sequenced mitogenomes, we designated curated reference sequences for 164 species. The resulting curated database provides a critical tool for resolving taxonomic ambiguities, clarifying phylogenetic relationships, and supporting forensic applications in wildlife conservation. Notably, seven of the 12 newly sequenced mitogenomes (58%) were derived from museum specimens, underscoring the enduring value of historical collections in modern conservation genomics [43], particularly for rare or elusive taxa such as the large-spotted civet, which was rediscovered after more than three decades [44].
Taxonomic clarification and remaining challenges
Our findings clarified species boundaries in several cases but also highlighted ongoing challenges in taxonomic uncertainty. For example, the mouse-deer Tragulus williamsoni was recognized as a distinct species by Meijaard and Groves [45], but considered a subspecies of T. kanchil by others [7]. We sequenced two historical specimens (KIZ75909 and KIZ75910) from Mengla, Xishuangbanna, Yunnan, which clustered with T. kanchil from Laos, supporting its subspecific status. However, as the type locality of williamsoni is in Thailand, further confirmation through sequencing of topotype material remains essential.
Taxonomic complexity is even greater in the genus Moschus. Sequences labeled as M. berezovskii and M. chrysogaster appeared in multiple clades (Fig 2B), suggesting cryptic speciation, misidentification, or gene flow. For instance, M. chrysogaster (JQ608470) may represent a misidentified individual or a hybrid [46]. Similarly, the mitogenome of M. leucogaster (NC_042604) from Lazi County, Tibet, clusters closely with M. fuscus, rather than with M. leucogaster from its type locality in Nepal [47], indicating likely misidentification. Genetic divergence of 5.9% within M. berezovskii, exceeding typical intraspecific thresholds, further underscores the need for integrative approaches to reassess species boundaries [48]. Additionally, paraphyly within M. fuscus and M. leucogaster add to the uncertainty, raising the possibility of incomplete lineage sorting or past introgression. Collectively, these findings underscore the necessity for a comprehensive taxonomic reassessment of Moschus. Resolving such ambiguities will require integrative taxonomic approaches that combine mitogenomic data with nuclear genomic analyses, morphology, ecology, and geographic sampling.
Forensic applications, future directions, and proposed best practices
The curated mitogenome database developed here, now encompassing 164 species and missing only five, represents a major step toward standardized species identification in China’s protected mammals. By uniting updated taxonomy, curated reference sequences, and data from historical specimens, it provides an authoritative resource that can be directly applied in biodiversity monitoring, forensic identification, and enforcement of conservation laws. However, the utility of this database will increase as it is expanded and refined, and its application should pioneer a more robust methodological standard for the field.
First, we strongly recommend the adoption of a phylogenetic framework for forensic species identification, rather than relying on simple genetic distance thresholds. As our results for Moschus demonstrate, genetic distance alone can be misleading in taxa with low interspecific divergence or complex evolutionary histories. Placing an unknown sample within a phylogenetic tree of curated reference sequences provides a far more robust and contextually supported identification. We propose that this phylogenetic approach should be established as a standard for forensic analysis, especially for legally protected species.
Second, our results demonstrate that reliance on single or geographically narrow reference sequences can be misleading for widespread species, in which intraspecific genetic structure can be substantial, and capturing this variation is critical. Incorporating mitogenomes from across the geographic ranges of widespread species will enable the assignment of confiscated specimens to specific populations or origins. Such resolution is vital for tracking illegal wildlife trade, as demonstrated in recent pangolin studies [49], and could be extended to other heavily trafficked groups such as musk deer and gorals.
Third, our database can serve as the foundation for expanding coverage to all mammals in China, not just those currently protected. Extending the database to species with ecological, economic, or cultural significance and eventually to all mammalian taxa would create a comprehensive national reference library. This expansion would enhance forensic capabilities by enabling identification of a broader range of trafficked species, including those not yet recognized as threatened but potentially at risk from overexploitation.
Fourth, our findings emphasize that the strength of forensic identification depends not only on genetic reference data but also on the robustness of the underlying taxonomy. In cases where taxonomy remains unresolved—such as Moschus and Naemorhedus—the reliability of forensic conclusions is diminished. Misaligned taxonomy can directly undermine the credibility of forensic reports in legal proceedings, highlighting the need for systematic taxonomic revision as a prerequisite for effective application of genetic tools. Moreover, in some groups, natural hybridization may occur, further complicating identification and requiring more nuanced frameworks for forensic reporting.
Finally, we acknowledge that mitochondrial genomes alone cannot fully resolve all cases of taxonomic or forensic ambiguity. Integration of nuclear and mitochondrial data, coupled with machine-learning approaches for species delimitation [48], which together will provide the most robust framework for both taxonomy and conservation enforcement.
Supporting information
S1 FigThe Maximum-likelihood trees of Budorcas taxicolor whitei (A) and Muntiacus vaginalis nigripes (B) based on mitochondrial genome dataset.Nodes with BS ≥ 95 are indicated by black dots, while those with BS below 95 are shown as numbers. Bold names are newly obtained sequences in this study. Red names are designated curated mitochondrial genome sequence for species based on type locality and phylogenetic relationships. Sequences from GenBank used in this study are named with their accession numbers.(TIF)
S1 TableThe updated list of State Key Protected Wild Mammals in China.(XLSX)
S1 DatasetThe alignment of curated mitochondrial genome sequences is available online (https://doi.org/10.57760/sciencedb.24175).(TXT)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lacher TE Jr, Davidson AD, Fleming TH, Gómez-Ruiz EP, Mc Cracken GF, Owen-Smith N, et al. The functional roles of mammals in ecosystems. Journal of Mammalogy. 2019;100(3):942–64. doi: 10.1093/jmammal/gyy 183 · doi ↗
- 2Banks-Leite C, Ewers RM, Folkard-Tapp H, Fraser A. Countering the effects of habitat loss, fragmentation, and degradation through habitat restoration. One Earth. 2020;3(6):672–6. doi: 10.1016/j.oneear.2020.11.016 · doi ↗
- 3Exposito-Alonso M, Booker TR, Czech L, Gillespie L, Hateley S, Kyriazis CC, et al. Genetic diversity loss in the Anthropocene. Science. 2022;377(6613):1431–5. doi: 10.1126/science.abn 5642 36137047 · doi ↗ · pubmed ↗
- 4Montràs-Janer T, Suggitt AJ, Fox R, Jönsson M, Martay B, Roy DB, et al. Anthropogenic climate and land-use change drive short- and long-term biodiversity shifts across taxa. Nat Ecol Evol. 2024;8(4):739–51. doi: 10.1038/s 41559-024-02326-7 38347088 PMC 11009105 · doi ↗ · pubmed ↗
- 5Wei F, Yang Q, Wu Y, Jiang X, Liu S, Hu Y. Catalogue of mammals in China. Acta Theriologica Sinica. 2025;45(1):1–16. doi: 10.16829/j.slxb.151039 · doi ↗
- 6Hu Y, Thapa A, Fan H, Ma T, Wu Q, Ma S, et al. Genomic evidence for two phylogenetic species and long-term population bottlenecks in red pandas. Sci Adv. 2020;6(9):eaax 5751. doi: 10.1126/sciadv.aax 5751 32133395 PMC 7043915 · doi ↗ · pubmed ↗
- 7Wei F. Taxonomy and distribution of mammals in China. Beijing: Science Press. 2022.
- 8Iyengar A. Forensic DNA analysis for animal protection and biodiversity conservation: A review. Journal for Nature Conservation. 2014;22(3):195–205. doi: 10.1016/j.jnc.2013.12.001 · doi ↗