AnaToc75 (Alr2269) mediates calcium uptake across the outer membrane in Anabaena sp. PCC 7120
Xiaoying Jiang, Hong Gao, Yanling Dong, Jianxin Tang, Shuiling Ji, Xudong Xu

TL;DR
This study shows that a cyanobacterial protein, Alr2269, functions as a calcium channel in the outer membrane, especially under low calcium conditions.
Contribution
The study reveals a new role for a Toc75 homolog in cyanobacteria as a calcium channel, challenging previous assumptions about its function.
Findings
Alr2269 is a specific channel for calcium uptake across the outer membrane in low-Ca2+ environments.
Inactivation of Alr2269 reduces growth under Ca2+-limiting conditions but not under low-Mg2+ conditions.
The protein is not essential for β-barrel protein insertion but is crucial for calcium transport.
Abstract
It has been a long-standing notion that cyanobacterial Omp85 proteins are essential because of their role in insertion of β-barrel proteins into the outer membrane (OM). Alr2269 from Anabaena sp. PCC 7120 is such an Omp85 protein, structurally related to the Toc75 channel of the translocon at the outer envelope membrane of chloroplasts but is actually non-essential to Anabaena under calcium-replete conditions. In a completely segregated alr2269-null mutant, a predicted S-layer protein, All3983, and many β-barrel proteins in the OM were upregulated compared with the wild type. By removal of each component from the medium, Ca2+ deficiency was identified to be the key environmental signal for upregulation of Pall3983-luxAB in the mutant. Assays of 45Ca2+ uptake in the wild type and the mutant indicated that Alr2269 is a relatively specific channel for transport of Ca2+ across the OM in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
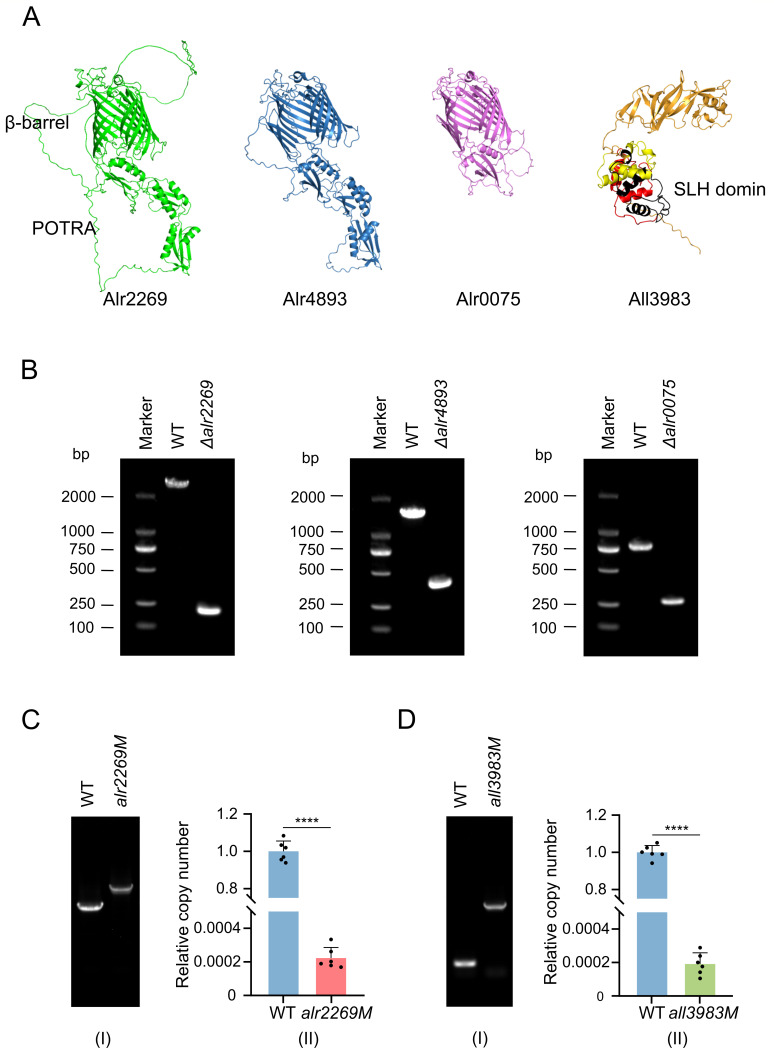 Fig 1
Fig 1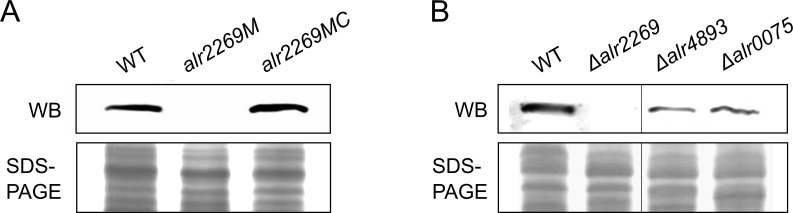 Fig 2
Fig 2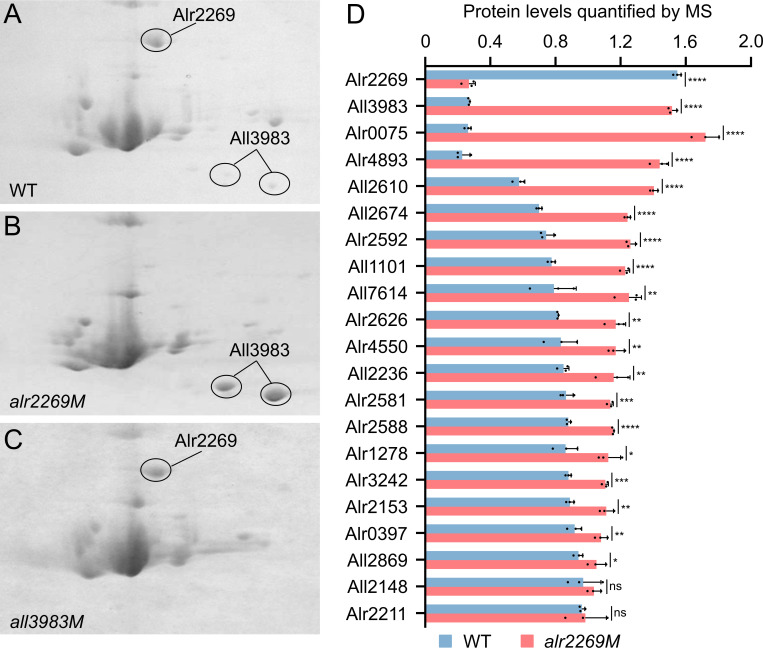 Fig 3
Fig 3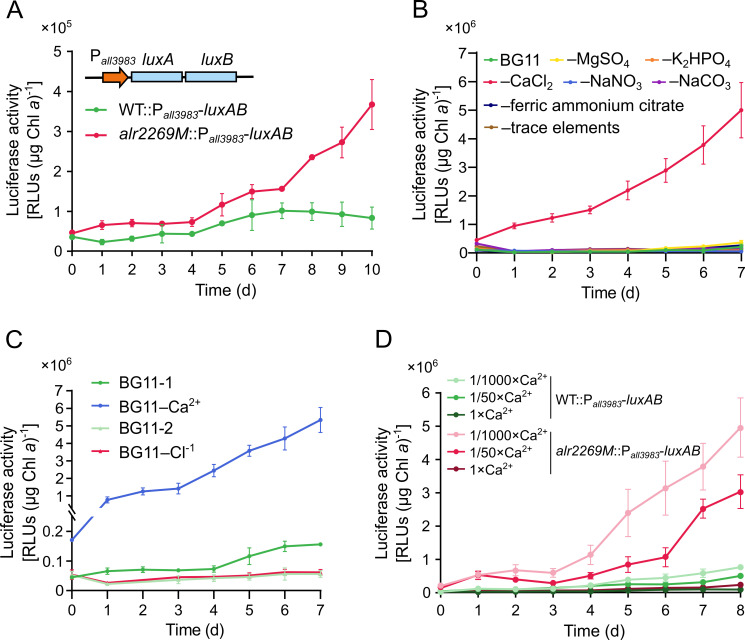 Fig 4
Fig 4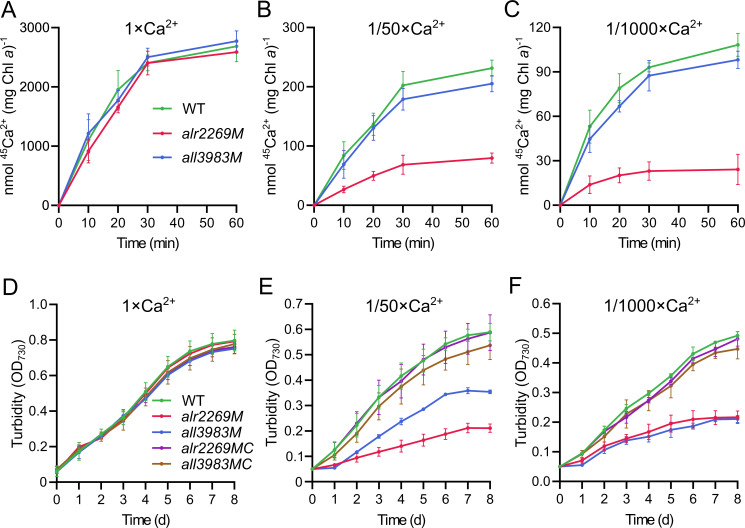 Fig 5
Fig 5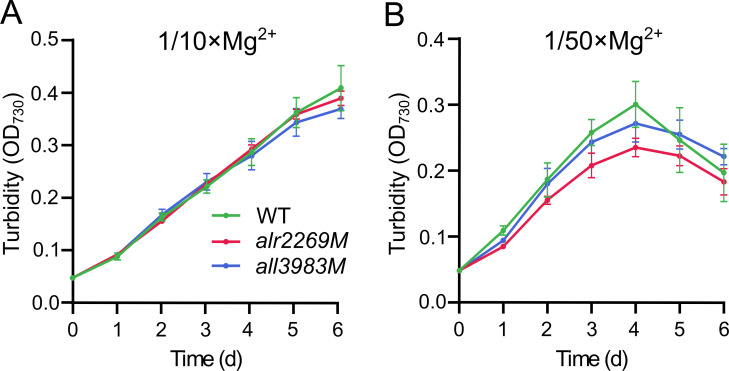 Fig 6
Fig 6- —National Key Research and Development Program of Chinahttp://dx.doi.org/10.13039/501100012166
- —National Natural Science Foundation of Chinahttp://dx.doi.org/10.13039/501100001809
- —National Natural Science Foundation of Chinahttp://dx.doi.org/10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhotosynthetic Processes and Mechanisms · Plant Stress Responses and Tolerance · Photoreceptor and optogenetics research
INTRODUCTION
Ca^2+^ is required for various biological processes, functioning as secondary messengers in cellular signaling (1, 2) and structural/catalytic cofactor for various enzymes (3–5). In cyanobacteria, Ca^2+^ plays an important role in photosynthesis (6, 7), motility (8, 9), cell differentiation (10), and stress responses (11, 12), and Ca^2+^ fluctuations synchronize cellular activities like circadian rhythms (13) and secondary metabolite production (14), crucial for ecological fitness and resource optimization. Furthermore, findings in Gram-negative bacteria (15) suggest that Ca^2+^ could play a structural role in the outer membrane of cyanobacteria as well, particularly in stabilization of the lipopolysaccharide layer and organization of S-layer proteins.
Although Ca^2+^ is so important to life activities, cytosolic free Ca^2+^ is maintained at submicromolar levels in cyanobacteria (10, 16, 17), about three orders of magnitude lower than the culture medium level, and may increase to 1–4 µM transiently in response to environmental stresses (16, 17). Similar differences between intra- and extracellular Ca^2+^ concentrations are found in other free-living bacteria (18, 19). For this reason, Ca^2+^ extrusion mechanisms (20) are much more extensively studied than uptake mechanisms (21–23). Regardless of efflux or influx, almost all Ca^2+^ transport systems hitherto reported in bacteria (24, 25), including those in cyanobacteria, are located in the cytoplasmic membrane, except the Ca^2+^ transporter across the outer membrane in Mycobacterium tuberculosis (26).
Alr2269 is an outer membrane protein in the heterocyst-forming cyanobacterium Anabaena sp. PCC 7120 (hereafter referred to as Anabaena 7120) and structurally related to Toc75 of the chloroplast outer envelope translocon complex (27), therefore dubbed AnaToc75. Alr2269 and homologs, such as Slr1227 (SynToc75) from Synechocystis sp. PCC 6803 (28, 29), belong to the Omp85 superfamily. Generally, Omp85 proteins in bacteria are involved in the insertion of β-barrel proteins into the outer membrane or translocation of proteins across the outer membrane (30). Cyanobacterial homologs of Toc75 are thought to be essential because both alr2269 and slr1227 mutants failed to be completely segregated (29, 31). One explanation is that they are required for integrating β-barrel proteins into the outer membrane ensuring membrane integrity and permeability (31, 32). Indeed, a partially segregated mutant of alr2269 showed altered sensitivity to erythromycin, SDS, lysozyme, and proteinase K and enhanced permeability to sucrose and glutamate. On the other hand, SynToc75 in reconstituted liposomes exhibits solute channel properties, allowing ions and small peptides to pass through the lipid layer (28). Thus, cyanobacterial homologs of Toc75 may act as ion channels as well.
In Anabaena 7120, there are two other Omp85 proteins, Alr4893 and Alr0075, homologous to Alr2269 (31). These Omp85 proteins (Fig. 1A) share a conserved β-barrel-type pore structure and the N-terminal POTRA (polypeptide translocation associated) domains (33) but differ in the pore diameter (1.3, 1.2, and 1.7 nm for Alr4893, Alr0075, and Alr2269, respectively) (31) and the number of POTRA domains (three for Alr4893 and Alr2269, one for Alr0075, see Fig. 1A). Like alr2269, alr4893 and alr0075 appeared to be essential to Anabaena 7120 because none of their insertion mutants were completely segregated (31, 32).
Predicted structures of Alr2269, Alr4893, Alr0075, and All3983 and the completely segregated Anabaena 7120 mutants of their encoding genes. PCR primers are indicated in Fig. S2 and listed in Table S1. (A) Predicted structures of Alr2269, Alr4893, Alr0075, and All3983. The three SLH domains (amino acid residue no. 21–84, 85–144, 146–210) are indicated in red, black, and yellow. (B) Electrophoretograms demonstrating PCR products amplified from genomes of Anabaena 7120 (WT) and the deletion mutants Δalr2269, Δalr4893, and Δalr0075. (C and D) PCR (I) and qPCR (II) examinations showing the complete segregation of the alr2269M and all3983M mutants. The relative copy numbers were calculated as 2-△△Ct values in Tables S4 and S5.
The lack of completely segregated mutants of Toc75 homolog genes in cyanobacteria may greatly impede the in-depth investigation of their functions because some phenotypes could be masked by the presence of the wild-type gene. Furthermore, the mutant strains would not be stable until complete segregation. For example, partially segregated alr2269 mutants showed very different sensitivities to lysozyme, proteinase K, SDS, and erythromycin in two independent reports (31, 32). In this study, we obtained the completely segregated alr2269-null mutant of Anabaena 7120, then we identified All3983, a predicted S-layer protein, as the Ca^2+^-responsive marker in the alr2269 background. Ca^2+^ uptake and low Ca^2+^ growth experiments showed the role of Alr2269 in transport of Ca^2+^ across the outer membrane. Unlike alr2269, the other two Omp85 protein genes were not required for growth in low Ca^2+^ medium. These results established Alr2269 (AnaToc75) as an important Ca^2+^ channel under Ca^2+^-limiting conditions, showing the role of a Toc75 homolog in ion transport.
RESULTS
Generation and characterization of the completely segregated alr2269 mutant
Previous studies reported that all three Omp85 protein genes are essential to Anabaena 7120 (31, 32). However, employing the CRISPR-Cpf1-based genome editing system (34, 35), we successfully deleted alr2269, alr4893, and alr0075 in Anabaena 7120, generating the completely segregated Δalr2269, Δalr4893, and Δalr0075 mutants (Fig. 1B; Fig. S1A). To confirm this result, we also generated an alr2269 insertion mutant, alr2269M, by inserting a chloramphenicol- and erythromycin-resistance cassette (C.CE2) into the Mlu I site of alr2269 (Fig. S2A). PCR detection showed a complete segregation of the insertion mutant. To strengthen the PCR examination result, we performed quantitative PCR to evaluate the wild-type copy number in the mutant. As shown in Fig. 1C, the wild-type form accounted for about 2 in 10,000 genome copies (virtually undetectable, the same level as the background), indicating that the alr2269M strain was indeed completely segregated. Then, we checked the Alr2269 protein in the mutants by Western blotting. Alr2269 was present in the wild type but not in the alr2269M (Fig. 2A) and Δalr2269 (Fig. 2B) mutants, and complementation of the alr2269M strain with alr2269 on a replicative plasmid restored the band of Alr2269 (Fig. 2A). Alr2269 was also detectable in the Δalr4893 and Δalr0075 mutants (Fig. 2B).
Western blot detection (WB) of Alr2269 in the wild type and mutants of Anabaena 7120. (A) Detection of Alr2269 in the wild type, alr2269M, and the complementation strain alr2269MC. (B) Detection of Alr2269 in the wild type, Δalr2269, Δalr4893, and Δalr0075.
Considering that Alr2269 is one of the three Omp85 proteins in Anabaena 7120, we investigated whether Alr2269 influences the insertion of β-barrel proteins into the outer membrane (OM). We isolated and purified the OM from the wild type (WT) and alr2269M and verified the purity of OM samples through SDS-PAGE analysis (Fig. S3A) and spectral scanning (Fig. S3B). Compared with total membranes, the purified OM showed much less protein bands and lost the characteristic chlorophyll a absorption peaks at 440 and 680 nm, retaining the carotenoid absorption peak at 487 nm.
We then subjected the OM samples of the WT and alr2269M to two-dimensional (2-D) gel electrophoresis. The 2-D patterns were quite similar to each other, except for the disappearance of one spot and the apparent accumulation of two others in alr2269M (Fig. 3A and B). Mass spectrometric analysis of these differentially expressed protein spots revealed that the missing protein was Alr2269, as expected for the insertion mutant, while the two accumulated spots corresponded to the same protein, All3983, a predicted S-layer protein. Its N-terminal amino acid no. 21-210 is 31.79% identical to the SLH domain of the surface layer (S-layer) protein SbpA of Lysinibacillus sphaericus (36) (Q9RER7) (E-value 2e-17).
*Proteomic analyses of outer membrane proteins in Anabaena 7120 (WT) and mutants. (A–C) Two-dimensional (2-D) gel electrophoretogram of outer membrane (OM) proteins of the WT, alr2269M, and all3983M. Protein spots were identified by using tandem mass spectrometry. (D) Quantitative analysis of β-barrel proteins and All3983 in the OM of WT and alr2269M (see Table S2 for a full list of OM proteins). All values are plotted as mean ± SD (n = 3). Asterisks denote significant differences by two-tailed Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001, ***P < 0.0001). ns, not significant.
To further evaluate the influence of Alr2269 on the OM protein profile, we performed TMT-labeled quantitative proteomic analysis of the OM samples. Among the mass spectrometry-detected proteins, 102 were identified as OM proteins using the functional annotation web server DAVID (37) and the protein subcellular localization prediction tool PSORTb v3.0.3 (38) (union of the two lists, see Table S2). β-Barrel proteins were predicted using the Deep TMHMM (39). The proteomic analysis showed that no β-barrel proteins disappeared in the OM of alr2269M compared to the WT; in contrast, most β-barrel proteins were upregulated or non-regulated in the mutant (Table S2, with some of the upregulated proteins shown in Fig. 3D). The proteomic data confirmed the significant upregulation of All3983 and the other two Omp85 proteins Alr4893 and Alr0075 in the alr2269M mutant relative to the WT.
Identification of all3983 as a Ca2+-responsive marker in the alr2269 mutant
In the absence of Alr2269, the abundance of All3983 significantly increased, suggesting a transcriptional or post-transcriptional regulation of all3983 in the mutant, so we employed the luciferase genes luxAB to monitor the promoter activity of all3983 in the WT and alr2269M. Pall3983-luxAB was integrated into the genome via single homologous recombination, generating strains WT::Pall3983-luxAB and alr2269M::Pall3983-luxAB.
After transfer of cells into the fresh BG11 medium, the transcription of all3983 gradually increased over time in the alr2269 mutant but remained nearly constant in the WT (Fig. 4A). The upregulation of all3983 could be the major reason for the accumulation of All3983 in alr2269M. We wondered if this can be enhanced by nutrient deficiency in the medium; therefore, we tested the response of Pall3983-luxAB in the alr2269 mutant to removal of each component from BG11 (Fig. 4B). Over 7 days, luciferase activity increased markedly only in the medium with CaCl_2_ omitted but remained at baseline levels when other components were removed. To differentiate between Ca^2+^ and Cl^-^, we replaced CaCl_2_ with Ca(NO_3_)2 or NaCl. Only lack of Ca^2+^ induced the upregulation of all3983 (Fig. 4C). Thus, the inducing factor should be lack of Ca^2+^ rather than Cl^-^. Furthermore, when WT::Pall3983-luxAB and alr2269M::Pall3983-luxAB were grown in BG11 with the [Ca^2+^] reduced to 1/50 or 1/1,000 of the standard level (240 µM), alr2269M::Pall3983-luxAB showed elevated expression of all3983 relative to the same strain in the standard BG11 medium (Fig. 4D). These findings strongly indicated that Alr2269 and All3983 are related to Ca^2+^ homeostasis.
Upregulation of all3983 in alr2269M in response to decrease of (Ca2+). (A) Expression of luxAB from the promoter of all3983 (Pall3983-luxAB) in the WT or alr2269M, as shown with the luciferase activity. The growth of Anabaena strains would elevate the pH of the medium, lowering the concentration of free Ca2+ (through the reaction with CO32-). The inset shows the structure of Pall3983-luxAB. (B) Responses of alr2269M::Pall3983-luxAB to removal of each component (indicated with the minus sign, for example, -MgSO4) from BG11. (C) Responses of alr2269M::Pall3983-luxAB to removal of Ca2+ (CaCl2 was replaced with NaCl) and Cl- (CaCl2 was replaced with Ca(NO3)2, and NaCl was removed). BG11-1 serves as the control for BG11-Ca2+, while BG11-2 serves as the control for BG11-Cl-. (D) Responses of WT::Pall3983-luxAB and alr2269M::Pall3983-luxAB to [Ca2+] in the medium. 1 × Ca2+ refers to BG11 (240 µM Ca2+); 1/50 × Ca2+, BG11 with 4.8 µM Ca2+; 1/1000×Ca2+, BG11 with 0.24 µM Ca2+.
We also generated an insertion mutant of all3983 (Fig. S2B), and the mutant all3983M was completely segregated as shown with PCR and quantitative PCR (Fig. 1D). 2-D gel electrophoresis of the OM proteins indicated that All3983 was absent while the abundance of Alr2269 remained unchanged in all3983M (Fig. 3A and C).
Role of Alr2269 in Ca2+ uptake
Given that all3983 was upregulated in response to Ca^2+^ depletion in the absence of Alr2269, and that both Alr2269 and All3983 are outer membrane proteins, we hypothesized that Alr2269 and All3983 may contribute to Ca^2+^ utilization. To test this, Ca^2+^ uptake assays were conducted in the WT and mutant strains, and their growth was compared under Ca^2+^-limiting conditions.
Ca^2+^ uptake was assayed using the ^45^Ca^2+^ isotope. As shown in Fig. 5A, no significant difference in ^45^Ca^2+^ uptake between strains was observed in standard BG11. However, when the [Ca^2+^] was reduced to 1/50 or 1/1,000 of the standard [Ca^2+^], alr2269M displayed a marked decrease in ^45^Ca^2+^ uptake compared with the WT, whereas all3983M was only slightly reduced in ^45^Ca^2+^ uptake (Fig. 5B and C). Notably, some Ca^2+^ uptake activities were still detectable in the absence of Alr2269.
Ca2+ uptake and utilization in Anabaena 7120 and derivative strains. (A–C) Time course of intracellular 45Ca2+ content in the WT, alr2269M, and all3983M strains after incubation in 1 × Ca2+, 1/50 × Ca2+, and 1/1,000 × Ca2+ media (45Ca2+ supplemented as 1/10,000 of total Ca2+). (D–F) Growth curves of the WT, alr2269M, all3983M, alr2269MC (alr2269M complemented with alr2269), and all3983MC (all3983M complemented with all3983) in 1 × Ca2+, 1/50 × Ca2+, and 1/1,000 × Ca2+ media.
The growth of these strains was also compared under Ca^2+^-limiting conditions. In BG11, alr2269M and all3983M grew as the wild type (Fig. 5D); however, in BG11 with 1/50 standard [Ca^2+^], the two mutants showed significantly reduced growth rates compared with the WT, with the growth retardation of alr2269M more pronounced (Fig. 5E); when [Ca^2+^] was reduced to 1/1,000, the two mutants showed comparably significant reduction in growth (Fig. 5F). To exclude the possibility of polar effects or second mutations, we complemented the mutants with the corresponding wild-type genes. The complemented strains alr2269MC and all3983MC grew as the wild type under the low Ca^2+^ conditions (Fig. 5E and F).
Because Ca^2+^ concentration is tightly controlled in bacterial cells by orchestration of efflux, uptake, and sequestration (such as binding to proteins) of Ca^2+^ (40), the intracellular free Ca^2+^ concentrations ([Ca^2+^]i) of the wild type and alr2269M would not show differences as large as that between Ca^2+^ uptake activities of the two strains. Even so, in Ca^2+^-free or 1/50 × Ca^2+^ medium, [Ca^2+^]i was significantly lowered in alr2269M compared with the wild type and restored to the wild type level in the complemented strain alr2269MC; in the 1×Ca^2+^ medium, alr2269MC showed even a higher [Ca^2+^]i than the wild type (Fig. S4).
We also examined the growth of the independent alr2269 mutant, △alr2269, under calcium-limiting conditions and found the same phenotype as alr2269M (Fig. S1B). However, the deletion mutants of the other two Omp85 protein genes, Δalr4893 and Δalr0075, grew as the wild type in the low Ca^2+^ medium.
Furthermore, we wondered whether Alr2269 is involved in utilization of Mg^2+^, Mg^2+^, and Ca^2+^ both belong to group IIA of the periodic table, thus are more similar to each other than other divalent cations. In BG11 with 1/10 of standard [Mg^2+^], alr2269M and all3983M grew as the wild type (Fig. 6A); at 1/50 of standard [Mg^2+^], alr2269M showed slightly slower growth than the wild type and all3983M, and all these strains started to decline in turbidity after 4 days (Fig. 6B). As the most abundant intracellular divalent cation, Mg^2+^ is involved in the synthesis of chlorophyll and many other biochemical reactions; thus, it was not feasible to further reduce the [Mg^2+^] in the medium. Considering 1/50 [Ca^2+^] (4.8 µM) and 1/50 [Mg^2+^] (6.0 µM) are at comparable levels, the remarkably different effects of Alr2269 on the growth under Ca^2+^- or Mg^2+^-limiting conditions ([Fig. 5E and 6B](#F5 F6)) suggest that Alr2269 is a relatively specific channel for Ca^2+^ uptake.
Growth of Anabaena 7120 and the alr2269M, all3983M mutants in BG11 with 1/10 × Mg2+ (A) or 1/50 × Mg2+ (B). The BG11 medium contains 300 µM MgSO4, while the 1/10 × Mg2+ and 1/50 × Mg2+ media contain 300 µM SO42- (MgSO4 and Na2SO4) and 30 µM or 6 µM Mg2+ (MgSO4).
DISCUSSION
Toc75 homologs in cyanobacteria, such as Slr1227 in Synechocystis and Alr2269 in Anabaena, have long been regarded as indispensable for cell viability, given their presumed role in outer membrane biogenesis. Previous studies, in which none of the mutants of these genes was completely segregated, lend support to this notion (29, 31).
In Synechocystis sp. PCC 6803, SynToc75 (Slr1227) is the only Omp85 protein. Under our conditions, an slr1227 mutant of Synechocystis was indeed partially segregated as previously reported (29). However, Anabaena 7120 possesses three Omp85 proteins, of which at least Alr2269 and Alr4893 are functionally redundant. In this study, we generated two independent alr2269 mutants of Anabaena 7120, and the mutants were both completely segregated. Fourfold evidence, including PCR, qPCR (Fig. 1C), Western blotting (Fig. 2), and 2-D gel electrophoresis (Fig. 3B), confirmed the complete absence of Alr2269 in alr2269M. These results clearly indicated that Alr2269 is non-essential for Anabaena 7120 in BG11. The discrepancy between our results and previous reports could be due to independent microevolution of Anabaena 7120 in laboratories. In our previous studies, over 300 mutations were identified between “PCC 7120” isolates from different laboratories and referred to these isolates as “substrains of PCC 7120”; occasionally, the same genotype may cause different phenotypes in these substrains (41). Perhaps, due to the divergence in genomic sequence, alr2269 is non-essential in the IHB substrain used in our studies but essential in the other substrains (31, 32).
To evaluate the influence of the inactivation of alr2269 on the OM protein profile, we conducted 2-D gel electrophoresis and TMT-labeled quantitative proteomic analyses to compare the mutant and the wild type. The 2-D gel electrophoresis showed the absence of Alr2269 and the accumulation of All3983 in the alr2269 mutant compared with the WT; the quantitative proteomic analysis revealed the upregulation of many more proteins, including All3983 and many β-barrel proteins in the mutant. No OM β-barrel protein disappeared in alr2269M. Apparently, Alr2269 is not essential for β-barrel protein integration into the OM. The upregulation of Alr4893, structurally similar to Alr2269, might have partially compensated for the inactivation of alr2269.
The accumulation of All3983 is one of the most prominent changes caused by inactivation of alr2269. Investigation of the effect of each component in the medium on the expression of all3983 led us to the finding that calcium deficiency induced the upregulation of all3983 in the alr2269 mutant. It is our hypothesis that there is a Ca^2+^-mediated signal transduction pathway between Alr2269 and the transcription of all3983. In other words, Alr2269 is probably involved in Ca^2+^ utilization, in particular transport across the OM. The Ca^2+^ uptake assays and the tests of growth in low-Ca^2+^ medium substantiated the role of Alr2269 in Ca^2+^ utilization, and the role of Alr2269 was confirmed by complementation of the alr2269 mutant. Because Alr2269 is not required for β-barrel protein integration into the OM, this protein itself should be the main channel for import of Ca^2+^ across the OM. The upregulated Alr4893 and many other β-barrel proteins may account for the remaining Ca^2+^ uptake in the alr2269 mutant (Fig. 5B and C). In the presence of Alr2269, Alr4893 and Alr0075 were not upregulated; therefore, they should have even less influence on the growth in low Ca^2+^ medium (Fig. S1B).
Although Mg^2+^ uptake was not assayed, the remarkable difference between the role of Alr2269 in growth under low-Mg^2+^ and low-Ca^2+^ conditions led us to propose that the Alr2269 channel is relatively specific for the import of Ca^2+^. The β-barrel architecture and conserved POTRA domains of Alr2269 align with features of the chloroplast Toc75 channel (27). These domains likely contribute to the gating mechanism that allows selective Ca^2+^ permeation.
Unlike Alr2269, All3983 is predicted to be an S-layer protein. In general, S-layer proteins are anchored to the cell surface through interactions between SLH domains and specific polysaccharide components in the cell wall, organized as two-dimensional crystalline arrays (42). As shown in Fig. 5B and C, All3983 is basically not involved in Ca^2+^ uptake. However, it may facilitate recruitment of Ca^2+^ to the cell surface and strengthen the stability of the OM under Ca^2+^-limiting conditions, thus is required for the growth in low-Ca^2+^ medium (Fig. 5E and F).
In summary, our results establish Alr2269 (AnaToc75) as the main channel protein across the OM of Anabaena for uptake of Ca^2+^ at low concentrations. In BG11 with standard [Ca^2+^], Alr2269 is dispensable (Fig. 5D) because diffusion through other β-barrel channels in the OM could supply sufficient Ca^2+^ to the intracellular space. In most freshwater environments, calcium is not a limiting factor for proliferation of cyanobacteria. However, free Ca^2+^ may be reduced to low concentrations when cyanobacteria grow to relatively high densities and the pH increases (due to the consumption of CO_2_). The evolution of a cyanobacterial Toc75 homolog into a Ca^2+^ channel provides an example of additional functions of Omp85 proteins in bacteria other than protein integration and translocation.
MATERIALS AND METHODS
Strains and culture conditions
Anabaena 7120 was obtained from the Freshwater Algae Culture Collection at the Institute of Hydrobiology, Chinese Academy of Sciences. Anabaena 7120 strains were maintained in BG11 medium (43) at 30°C in the light of 10–20 μE m^−2^ s^−1^ without agitation. When necessary, antibiotics were added to the medium as follows: erythromycin at 5 µg mL^−1^, spectinomycin at 10 µg mL^−1^, and neomycin at 20 µg mL^−1^.
For generation of growth curves at different Ca^2+^ concentrations, Anabaena cells were cultured in transparent plastic bottles, with illumination of 30 μE m^−2^ s^−1^ from both upper and lower sides, manually agitated twice a day. Cells washed with Ca^2+^-free BG11 and pre-cultured in BG11 with different Ca^2+^ concentrations were inoculated into the same medium at an initial optical density (OD_730_) of 0.05, and the OD_730_ values were measured daily and plotted against time. For the growth in low Ca^2+^ BG11, illumination from both sides with manual agitation produced more consistent and regular growth curves than illumination only from the upper side with constant agitation on a rotary shaker.
Construction of plasmids and Anabaena strains
Molecular cloning was performed following standard protocols or manufacturer’s instructions. DNA fragments amplified by polymerase chain reactions (PCRs) were confirmed by sequencing after cloning. DNA ligations were conducted either by use of T4 DNA ligase (ligation of compatible dsDNA ends) or 2× MultiF Seamless Assembly Mix (ABclonal, Wuhan, China) (in vitro homologous recombination). Plasmids were introduced into the Anabaena 7120 strains by conjugation (44). Mutants were generated by deleting a stretch of sequence within a gene using the Cpf1-based genome editing system (35), or by interrupting a gene with an antibiotic resistance cassette using the strategy of sacB-based positive selection (45). Complete segregation of mutants was validated by PCR. Construction of plasmids and Anabaena strains, and sequences of PCR primers, are described in detail in Table S1.
Quantitative PCR (qPCR) analyses
Genomic DNA (gDNA) was extracted and purified using a mini-prep method (45). qPCR was performed according to the core principles of the MIQE guidelines (46), using SYBR Green Realtime PCR Master Mix (Toyobo, Osaka, Japan), in 96-well plates on a Bio-Rad CFX96 Real-Time PCR System (Bio-Rad Laboratories, Hercules, USA). Twenty microliters of reaction mixtures contained 10 µL of master mix, 6 pmol of each primer, 10 ng of gDNA, and nuclease-free water. PCRs initiated with 95°C for 3 min, followed by 40 cycles of: 95°C for 15 s, 60°C for 15 s, and 72°C for 15 s. Three no-template controls per plate were included. A pooled gDNA standard (equimolar mix of all samples) was serially diluted twofold for six concentration points and used to generate the standard curve, with each dilution run in triplicate. Amplification efficiency, standard curve linearity R^2^ and single peak melting temperature for each pair of primers are summarized in Table S3. Primers were designed based on unique sequences in the genome, and the specificity of a primer pair was demonstrated by the single band of agarose electrophoresis and the single peak of melting curve. Relative abundance of residual wild-type alleles was normalized with rnpB as the reference gene in the chromosomal DNA to correct variations between samples (47), whose efficiency differed by <5% from target genes. Data were analyzed using the 2^−ΔΔ^^C^^T^ method (48), and raw C_t_ values and calculated results are listed in Tables S4 and S5.
Western blot analysis
Anabaena 7120 cells were washed with 20 mM Tris-HCl (pH 8.0) containing 1 mM PMSF and resuspended in the same buffer. Cells were broken using a French press (Scientz, Ningbo, China) at a pressure of 15 MPa, followed by centrifugation at 12,000×g for 15 min at 4°C. The resulting supernatant was collected as total cell extracts for Western blot analysis. Protein samples were mixed with loading buffer (Zomabio, Beijing, China) and boiled for 5 min. Protein concentrations were determined using the Bradford Protein Assay Kit (CWBio, Taizhou, China). Equal amounts of proteins (20 µg per lane) were resolved by 12% SDS-PAGE and electro-blotted onto nitrocellulose (NC) membranes (MilliporeSigma, Burlington, USA). NC membranes were blocked with 5% skim milk in TBS-1 (25 mM Tris-HCl, 500 mM NaCl, pH 7.5) for 4 h at room temperature, then incubated overnight at 4°C with anti-Alr2269 rabbit antiserum (Qiwei Yicheng, Beijing, China; 1:1000 dilution in 1% skim milk). After washing three times with TBS-1, NC membranes were incubated with HRP-conjugated goat anti-rabbit IgG secondary antibody (Thermo Scientific, Waltham, USA; 1:5000 in 1% skim milk) for 2 h, then washed three times with TBST (25 mM Tris-HCl, 500 mM NaCl, 0.05% Tween-20, pH 7.5). Signals were developed using Omni-ECL Substrate (YamayBio, Shanghai, China) for 1 min and detected with ImageQuant LAS 4000 mini system (GE Healthcare, Chicago, USA).
Isolation of the outer membrane of Anabaena 7120
Outer membrane isolation was performed by aqueous two-phase partitioning followed by sucrose density gradient ultracentrifugation as described (49) with modifications. Briefly, cells from 2 L of culture (OD_730_ ≈ 1.2) were harvested by centrifugation and washed and resuspended with 20 mM potassium phosphate buffer (pH 7.8) and 1% protease inhibitor cocktail (APExBIO, Houston, USA), then stored at −70°C. Frozen cells were disrupted by vortexing with glass beads, the cell suspension was centrifuged at 3,300×g for 10 min to pellet debris, and the supernatant was collected. Total membranes were collected by ultracentrifugation (Optima L-100 XP, SW 41 Ti, Beckman Coulter, Brea, USA) at 103,000×g for 30 min. The membrane suspension was subjected to aqueous two-phase partitioning using a system containing 6.5% (w/w) dextran T-500, 6.5% (w/w) PEG3350, 0.25 M sucrose, and 5 mM potassium phosphate (pH 7.8). The second partitioning step was performed with a system containing 7.2% (w/w) dextran T-500, 7.2% (w/w) PEG3350, 0.25 M sucrose, and 5 mM potassium phosphate (pH 7.8). The outer membrane was further purified by two rounds of sucrose density gradient ultracentrifugation at 197,000×g for 6 h at 4°C. Purity of the isolated outer membrane was analyzed by SDS-PAGE and spectral scanning with UV2450 UV spectrophotometer (Shimadzu, Kyoto, Japan).
Two-dimensional gel electrophoresis
Total protein samples were dissolved in the solubilization buffer (9 M urea, 4% CHAPS, 2% IPG buffer, 40 mM DTT). An appropriate amount of sample was mixed with the rehydration buffer (8 M urea, 2% CHAPS, 15 mM DTT, 0.5% IPG buffer) to a total volume of 250 µL containing 270 µg proteins and loaded onto immobilized pH gradient (IPG) strips. Isoelectric focusing was performed as follows: 50 V for 12 h (rehydration), 200 V for 1 h, 500 V for 1 h, 1,000 V for 1 h, 2,000 V for 2 h, 3,500 V for 17 h. After isoelectric focusing, IPG strips were equilibrated by shaking for 15 min in 10-mL equilibration buffer I (0.05 M Tris-HCl, pH 8.8, 6 M urea, 30% [w/v] glycerol, 2% [w/v] SDS, 0.002% [w/v] bromophenol blue, 64.8 mM DTT) followed by 15 min in 10 mL equilibration buffer II (0.05 M Tris-HCl, pH 8.8, 6 M urea, 30% [w/v] glycerol, 2% [w/v] SDS, 0.002% [w/v] bromophenol blue, 0.216 M iodoacetamide). Strips were then rinsed in deionized water and applied to 12% SDS-PAGE gel for second dimension electrophoresis at 20 mA for 0.5 h, followed by 35 mA for 4.5 h. Gels were stained with Coomassie Brilliant Blue.
TMT-labeled quantitative proteomic analysis
Outer membrane samples (with approximately 100 µg proteins) were subjected to ultrasonic treatment on ice in the lysis buffer (8 M urea, 1% protease inhibitor cocktail). After centrifugation at 12,000×g at 4°C for 10 min, the protein solutions were collected and treated with 5 mM dithiothreitol at 56°C, followed by alkylation with 11 mM iodoacetamide in the dark at room temperature. The solutions were diluted with 100 mM Tetraethylammonium bromide (TEAB) solution, lowering the urea concentration to below 2 M. Proteins were digested with trypsin at 37°C (1:50 [w/w], overnight; 1:100 [w/w] for 4 h), and the resulting peptides were desalted using a Strata X C18 SPE column (Phenomenex, Torrance, USA) and vacuum-dried, then dissolved in 0.5 M TEAB buffer and processed with the TMT reagent kit (Thermo Fisher Scientific, Waltham, USA) according to manufacturer′s protocol. After labeling, peptides were analyzed on an EASY-nLC 1000 UPLC system at a constant flow rate of 400 nL/min for NSI source detection, followed by tandem mass spectrometry (MS/MS) on a Q Exactive Plus (Thermo Scientific, Waltham, USA) coupled with the UPLC system. MS/MS data were processed using the MaxQuant search engine (v.1.5.2.8) (50). Protein abundance was quantified based on peptide intensity values. Intensity values of each peptide were normalized across the six samples (3 WT + 3 alr2269M) to the average value. The peptide quantification values were then normalized to the median value of each sample. Unique peptides for each protein were identified, and protein quantification values were derived from the median of non-zero unique peptide values. The relative quantification ratio (alr2269M / WT) was calculated by comparing the average values of each protein between WT and alr2269M (three biological repeats). A Student’s t-test was performed on the Log_2_ (alr2269M / WT) values to analyze statistical significance.
Ca2+ uptake assays
Ca^2+^ uptake assay was conducted as described (51) with modifications. Cyanobacterial cells were washed three times with Ca^2+^-free BG11 to remove extracellular Ca^2+^, re-suspended in Ca^2+^-free BG11, and cultured for 5–7 days. Ca^2+^ uptake was initiated by re-suspending the Ca^2+^-depleted cells in BG11 containing different concentrations of ^45^Ca^2+^. At various incubation times (0, 10, 20, 30, 60 min), 1-mL aliquots were vacuum-filtered through 0.22-µm NC membranes pre-soaked in deionized water and washed three times with TBS-2 buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.5). The NC membranes with cells were transferred to scintillation vials containing 2 mL high-efficiency mineral oil scintillator and subjected to counting with the MicroBeta^2^ scintillation counter (PerkinElmer, Waltham, USA). A standard curve of scintillation counts per minute (cpm) against ^45^Ca^2+^ concentrations was established by measuring the radioactivity of serially diluted ^45^Ca^2+^ solutions, and ^45^Ca^2+^ concentrations of samples were interpolated from the standard curve. Chlorophyll a was extracted with methanol in the dark and determined based on the absorption coefficient of Chl a at OD_664_ (52). ^45^Ca^2+^ imported into cells was calculated based on three biological replicates and expressed in nmol ^45^Ca^2+^ (mg Chl a)^−1^.
Measurements of intracellular free Ca2+ using Fura-4 AM
Intracellular free calcium concentrations [Ca^2+^]i of Anabaena 7120 and derivative strains were evaluated using Fura-4 AM as described (53) with modifications. Forty microliters of Anabaena cells was collected by centrifugation, washed three times with Ca^2+^-free BG11 and allowed to grow in the same medium (to deplete stored Ca^2+^) for 5 days. The cells were then collected and washed three times with buffer A (50 mM Tris-HCl, 100 mM KCl, 1 mM MgCl₂, pH 7.5) and re-suspended in 1 mL of 0.12 M Tris-HCl (pH 8.0) containing 0.2 mM EDTA, shaken at 30°C for 2 min, and supplemented with MgCl₂ to a final concentration of 1 mM. Subsequently, the cells were washed three times with buffer A, re-suspended in 1 mL of buffer A, and loaded with 2.5 µM Fura-4 AM (Beyotime, Shanghai, China) in the dark at 30°C for 1 h. Again, the cells were washed three times and suspended in 1 mL of buffer A. Then, 500 µL of Fura-4 AM-loaded cells was added to 20 mL of Ca^2+^-free BG11, or 1 × Ca^2+^ BG11, or 1/50 × Ca^2+^ BG11 and incubated for 1 h, collected and re-suspended in 500 µL of buffer A. Fluorescence was recorded using a SpectraMax i3x microplate reader (Molecular Devices, Sunnyvale, USA) with excitation at 488 nm and emission at 520 nm. After permeabilization of cells with 25 µM gramicidin (MedChemExpress, Monmouth Junction, USA), Fmax and Fmin were determined by adding 1 mM CaCl₂ and 10 mM EGTA, respectively. [Ca^2+^]i was calculated using the formula: [Ca^2+^]i = K_d_ × (F − Fmin) / (Fmax − F). One-way ANOVA analysis followed by LSD post hoc tests was performed to determine significant differences in [Ca^2+^]i between groups.
Determination of bacterial luciferase activity
Luciferase activity was measured as described previously (54). Briefly, 1 mL of cells was mixed with 100 µL of 0.1% (v/v) n-decanol with 20 mg mL^−1^ BSA. The mixture was placed in an ATP photometer (Berthold, Gosheim, Germany) to measure relative light units (RLUs) for 60 s. In parallel, 1.2 mL of the cell suspension was centrifuged at 12,000×g for 5 min, and the pellet was re-suspended in 1.2 mL methanol for extraction of chlorophyll a. Luciferase activity was expressed as RLUs (μg Chl a)^−1^.
Bioinformatic analyses
Identification of outer membrane proteins was assisted by using the functional annotation web server DAVID (37) and the protein subcellular localization prediction tool PSORTb v3.0.3 (38). Structures of outer membrane proteins were downloaded from the AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/) and visualized using PyMOL software (Version 1.7, Schrödinger, LLC.; https://pymol.org). β-Barrel structures were also predicted using the Deep TMHMM (39).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Berridge MJ, Bootman MD, Roderick HL. 2003. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4:517–529. doi:10.1038/nrm 115512838335 · doi ↗ · pubmed ↗
- 2Newton AC, Bootman MD, Scott JD. 2016. Second messengers. Cold Spring Harb Perspect Biol 8:a 005926. doi:10.1101/cshperspect.a 00592627481708 PMC 4968160 · doi ↗ · pubmed ↗
- 3Ubarretxena-Belandia I, Boots JW, Verheij HM, Dekker N. 1998. Role of the cofactor calcium in the activation of outer membrane phospholipase A. Biochemistry 37:16011–16018. doi:10.1021/bi 98141819843408 · doi ↗ · pubmed ↗
- 4Ahvazi B, Boeshans KM, Idler W, Baxa U, Steinert PM. 2003. Roles of calcium ions in the activation and activity of the transglutaminase 3 enzyme. J Biol Chem 278:23834–23841. doi:10.1074/jbc.M 30116220012679341 · doi ↗ · pubmed ↗
- 5Karkare S, Yousafzai F, Mitchenall LA, Maxwell A. 2012. The role of Ca 2+ in the activity of Mycobacterium tuberculosis DNA gyrase. Nucleic Acids Res 40:9774–9787. doi:10.1093/nar/gks 70422844097 PMC 3479174 · doi ↗ · pubmed ↗
- 6Najafpour MM, Govindjee. 2011. Oxygen evolving complex in photosystem II: better than excellent. Dalton Trans 40:9076. doi:10.1039/c 1dt 10746 a 21735020 · doi ↗ · pubmed ↗
- 7Walter J, Selim KA, Leganés F, Fernández-Piñas F, Vothknecht UC, Forchhammer K, Aro EM, Gollan PJ. 2019. A novel Ca 2+-binding protein influences photosynthetic electron transport in Anabaena sp. PCC 7120. Biochim Biophys Acta 1860:519–532. doi:10.1016/j.bbabio.2019.04.00731034800 · doi ↗ · pubmed ↗
- 8Pitta TP, Sherwood EE, Kobel AM, Berg HC. 1997. Calcium is required for swimming by the nonflagellated cyanobacterium Synechococcus strain WH 8113. J Bacteriol 179:2524–2528. doi:10.1128/jb.179.8.2524-2528.19979098048 PMC 178999 · doi ↗ · pubmed ↗