Neuartige Variante des NCSTN‐Gens bei einer Frau mit familiärer Hidradenitis suppurativa
Conrad Hempel, Sonja Grunewald, Till Mittank‐Weidner, Jan‐Christoph Simon, Franziska Schnabel, Robin‐Tobias Jauss, Viktor Schnabel

Abstract
Click any figure to enlarge with its caption.
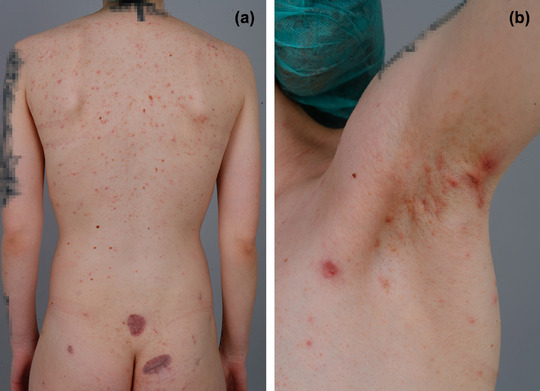 Figure 1
Figure 1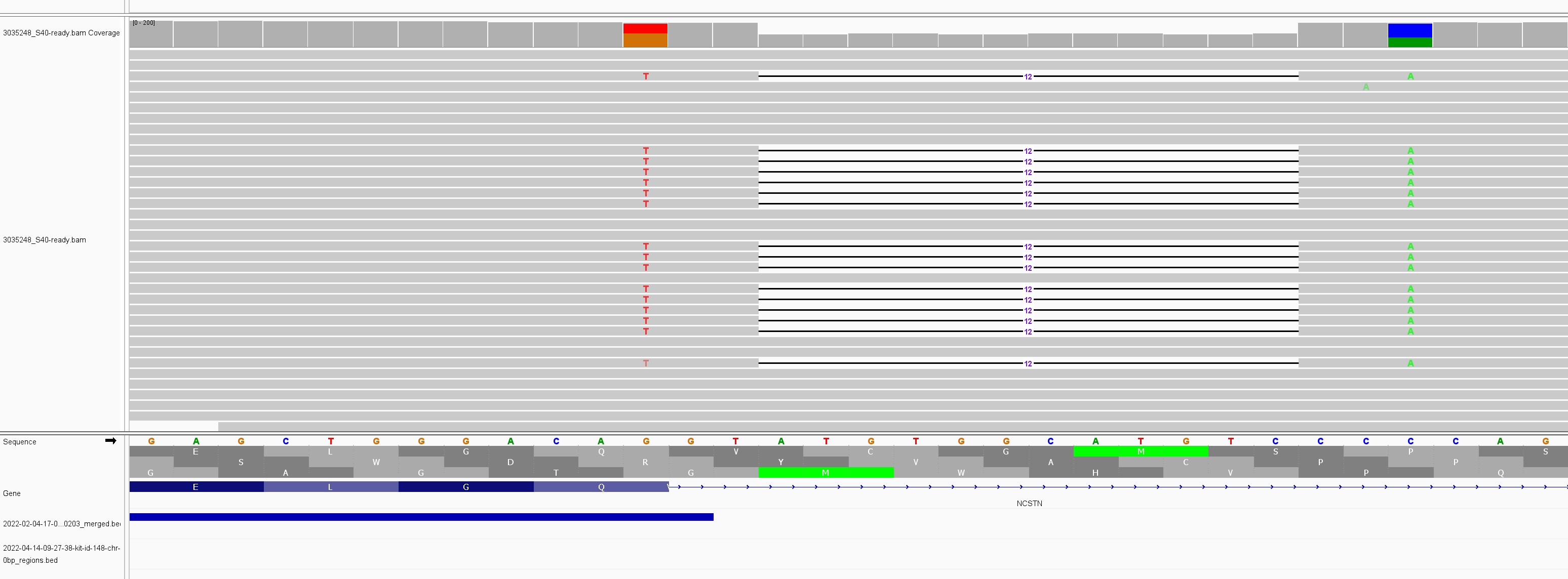 Figure 2
Figure 2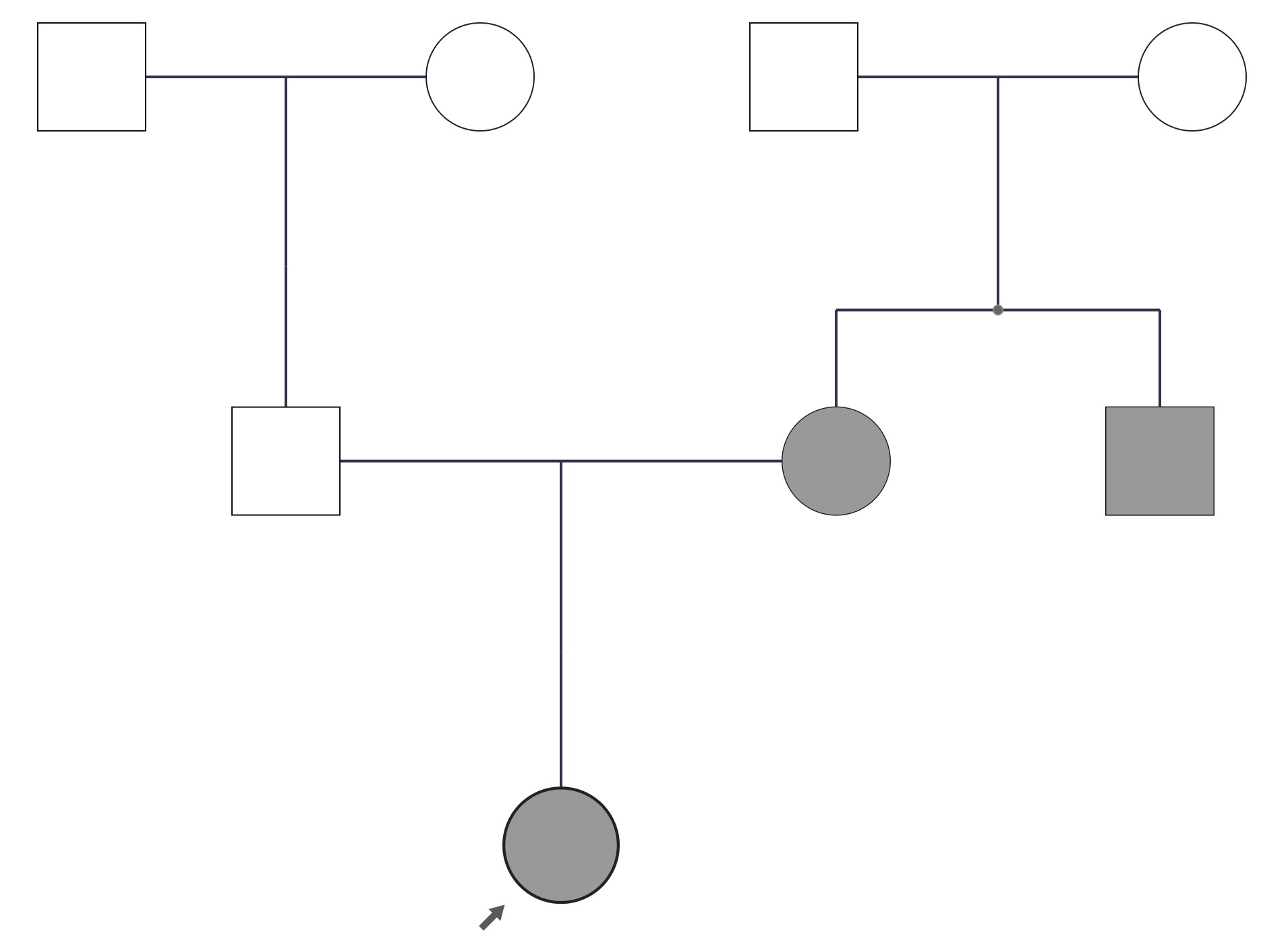 Figure 3
Figure 3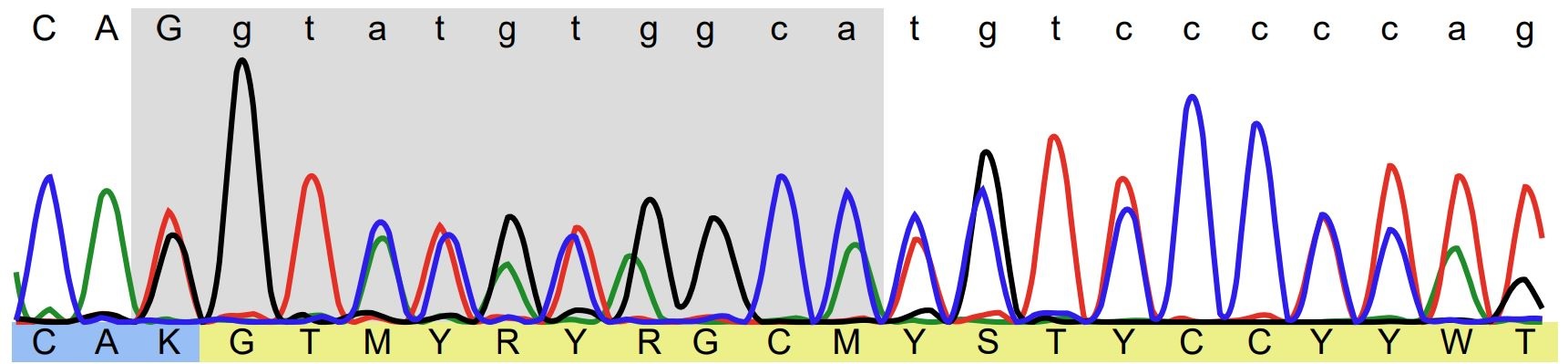 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHidradenitis Suppurativa and Treatments · Colorectal and Anal Carcinomas · Chemotherapy-related skin toxicity
Sehr geehrte Herausgeber,
Eine 27‐jährige kaukasische Frau mit einer vierjährigen Vorgeschichte einer Hidradenitis suppurativa stellte sich in unserer Ambulanz vor. Sie berichtete von wiederholten umfangreichen chirurgischen Eingriffen mit Spaltung von Hautabszessen in der Achselhöhle und in der Leiste, die interessanterweise auch an ungewöhnlichen Stellen wie Rumpf und Oberschenkeln auftraten. Zuvor wurde sie mit intermittierenden Antibiotika, Isotretinoin und einem hormonellen Kontrazeptivum behandelt. Zusätzlich wurde eine Therapie mit Tumornekrosefaktor‐alpha‐Antikörpern für 6 Monate sowie zuletzt eine Anti‐IL17‐Therapie für 3 Monate durchgeführt. Alle Medikamente sind aufgrund von Nebenwirkungen oder Unwirksamkeit abgesetzt worden.
Sie klagte vor allem über starke Schmerzen infolge der wiederkehrenden Abszesse, die zu einer Arbeitsunfähigkeit führten. Sie verneinte Nikotinkonsum und hat einen BMI von 18,5. Aufgrund einer begleitenden Depression und Angststörung befindet sie sich in psychologischer Betreuung und hat einen DLQI‐Wert von 29.
Die dermatologische Untersuchung ergab zahlreiche entzündliche Papeln, Komedonen und Pusteln, die über den ganzen Körper verteilt waren, wobei Gesicht, Rumpf und Oberschenkel am stärksten betroffen waren. Abszesse mit einem Durchmesser von bis zu 2 cm fanden sich in beiden Achselhöhlen sowie im Scham‐ und Gesäßbereich (Abbildung 1a,b), entsprechend einem Hurley‐Grad 2. Interessanterweise wiesen ihre Mutter und ihr Onkel mütterlicherseits ebenfalls intermittierende schmerzhafte Abszesse auf, die jedoch weniger stark ausgeprägt waren (Abbildung S1, Online‐Supplement).
Aufgrund der positiven Familienanamnese führten wir eine Whole‐Exom‐Sequenzierung mit dem TWIST Human Core Exome Kit (TWIST Bioscience, San Francisco, USA) auf einem Illumina NovaSeq6000‐Sequenzierer (Illumina, San Diego, USA) durch. Die Auswertung erfolgte mit der browserbasierten Genomiksoftware Varvis (Limbus Medical Technologies GmbH, Rostock, Deutschland). Hierbei zeigte sich eine heterozygote trunkierende Variante (NM_015331.3:c.1101_1101+17delinsTGTCCA, p.(Gln367Hisfs*6)) im NCSTN‐Gen (Abbildung S2, Online‐Supplement). Die mittlere Abdeckung des NCSTN‐Gens betrug 79,49x, und die Variante wurde in mehr als 50 Reads mit einer Qualitätsbewertung von 1918 (GATK HaplotypeCaller) und ohne diskrepante Reads in diesem Bereich nachgewiesen. Zur Bestätigung wurde eine Sanger‐Sequenzierung durchgeführt (Abbildung S3, Online‐Supplement). Die Variante führt zu einem Frameshift mit verkürzter Proteintranslation, wurde bislang jedoch weder in Varianten‐Datenbanken (HGMD, Decipher, ClinVar) noch in der Literatur beschrieben. In der allgemeinen Referenzdatenbank für Populationsfrequenzen (gnomAD) die Sequenzdaten von mehr als 1 150 000 europäischen (nicht finnischen) Kontrollpersonen an der Variantenposition enthält, ist die Variante nicht gelistet. Nach der ACMG‐Klassifikation wurde die Variante als pathogen eingestuft (angewandte Kriterien: PVS1, PM2_SUP, PP4).1 Leider standen keine weiteren Familienmitglieder für eine Segregationsanalyse zur Verfügung.
Hidradenitis suppurativa (HS), auch bekannt als Acne inversa, ist eine chronische, entzündliche und rezidivierende Erkrankung der Haarfollikel, die sich typischerweise durch schmerzhafte, entzündete Läsionen äußert. Am häufigsten sind die axilläre, inguinale und anogenitale Region betroffen.2 Die Prävalenz in Europa liegt bei etwa 1%.2 Das typische Erkrankungsalter bei Erstmanifestation liegt zwischen 20 und 40 Jahren.3 Etwa 40% der Fälle lassen sich als familiäre Form einordnen, neben syndromalen und sporadischen Varianten.3 Die familiäre HS ist in der Regel durch schwerere Symptome und einen früheren Krankheitsbeginn gekennzeichnet.3 In der Literatur wurden bisher mindestens vier verschiedene Gene aus dem y‐Sekretase‐Komplex als ursächlich für HS beschrieben, darunter NCSTN, PSENEN, PSEN1 und APH1B.4, 5 NCSTN kodiert für Nicastrin, ein Typ‐1‐Transmembran‐Glykoprotein des y‐Sekretase‐Komplexes, das eine zentrale Rolle im Notch‐Signalweg spielt und antiproliferative und differenzierungsfördernde Effekte in menschlichen Keratinozyten steuert. Die Ausschaltung von NCSTN führt zu einer verstärkten Proliferation und verminderten Differenzierung von Keratinozyten, die in erster Linie durch den Notch‐ und PIK‐AKT‐Signalweg vermittelt wird.6 Bisher wurden 52 Varianten von NCSTN bei HS beschrieben.5 Eine monogene Variante von NCSTN ist mit einem früheren Auftreten und einer atypischen Präsentation an ungewöhnlichen Hautstellen wie dem Rumpf verbunden. Darüber hinaus unterscheiden ein niedriges Körpergewicht und eine über den Krankheitsverlauf erhöhte Wahrscheinlichkeit einer Behandlung mit biologischen Therapien diese genetische Varianten von der klassischen HS, wie dieser Fall bestätigt.7 Das Zusammenspiel zwischen genetischen Faktoren und der Dysregulation von Immunmediatoren wie TNF‐α, IL‐1β, IL‐17 und IL‐12/23 trägt zum chronisch‐entzündlichen Charakter der HS bei.3 Potenzielle therapeutische Ziele umfassen daher Anti‐TNF‐α‐, Anti‐IL‐1α‐, Anti‐IL‐17‐ und Anti‐IL‐12/23‐Therapien sowie JAK‐Inhibitoren.
Dieser Fall untermauert die funktionelle Bedeutung von NCSTN‐Varianten in der Pathogenese der familiären HS, die bisher vor allem in asiatischen Populationen beschrieben wurde.8, 9
Bei unserer Patientin haben wir eine neue trunkierende NCSTN‐Variante beschrieben, die zur Entwicklung von HS führt. Interessanterweise konnten keine umweltbedingten Risikofaktoren, wie Adipositas oder Rauchen, festgestellt werden. Klinisch gehört der Phänotyp zum selteneren „follikulären“ Subtyp der HS, der von Canoui‐Poitrine et al. als einer von drei Subtypen beschrieben wurde.10 Dieser ist hauptsächlich durch follikuläre Läsionen wie Komedonen, epidermale Zysten sowie dem Auftreten von Sinus pilonidalis und Akne vulgaris gekennzeichnet und zeigt einen schwereren Krankheitsverlauf als die reguläre axilläre/inguinale Variante. Es muss darauf hingewiesen werden, dass die Komedonen unserer Patientin nicht die alleinige Ursache für die schwere, destruktive Entzündung sein können. Angesichts des genetischen Hintergrunds liefert dieser Bericht weitere Hinweise darauf, dass HS nicht länger als einfache „Erkrankung der Follikelokklusion“ betrachtet werden kann. Die bedeutende Rolle der Entzündung sowie der angeborenen und adaptiven Immunität bei familiärer HS kann nicht hoch genug eingeschätzt werden und wirft die Frage auf, ob die Follikelokklusion ein primäres oder sekundäres Phänomen in der Pathogenese der Erkrankung ist.
Unser Fallbericht erweitert das genetische Spektrum, das der HS zugrunde liegt. Zusammen mit dem klinischen Phänotyp liefert er wertvolle Informationen für weitere Studien zur Therapie und zur Entwicklung von Überwachungsprogrammen für betroffene Familien.
DANKSAGUNG
Open access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.
INTERESSENKONFLIKT
Keiner.
Supporting information
Supplementary information
Supplementary information
Supplementary information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Richards S , Aziz N , Bale S , et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424.25741868 10.1038/gim.2015.30PMC 4544753 · doi ↗ · pubmed ↗
- 2Zouboulis CC , Desai N , Emtestam L , et al. European S 1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29(4):619‐644.25640693 10.1111/jdv.12966 · doi ↗ · pubmed ↗
- 3Chu YL , Yu S . Hidradenitis Suppurativa: An Understanding of Genetic Factors and Treatment. Biomedicines. 2024;12(2):338.38397941 10.3390/biomedicines 12020338 PMC 10886623 · doi ↗ · pubmed ↗
- 4Pink AE , Simpson MA , Desai N , et al. Mutations in the γ‐secretase genes NCSTN, PSENEN, and PSEN 1 underlie rare forms of hidradenitis suppurativa (acne inversa). J Invest Dermatol. 2012;132(10):2459‐2461.22622421 10.1038/jid.2012.162 · doi ↗ · pubmed ↗
- 5Ratnamala U , Jain N , Jhala D , et al. An Updated Mutation Spectrum of the γ‐Secretase Complex: Novel NCSTN Gene Mutation in an Indian Family with Hidradenitis Suppurativa and Acne Conglobata. Indian J Dermatol. 2023;68(2):141.37275792 10.4103/ijd.ijd_995_21PMC 10238988 · doi ↗ · pubmed ↗
- 6Xiao X , He Y , Li C , et al. Nicastrin mutations in familial acne inversa impact keratinocyte proliferation and differentiation through the Notch and phosphoinositide 3‐kinase/AKT signalling pathways. Br J Dermatol. 2016;174(3):522‐532.26473517 10.1111/bjd.14223 · doi ↗ · pubmed ↗
- 7Mintoff D , Pace NP , Borg I . NCSTN In‐Frame Deletion in Maltese Patients With Hidradenitis Suppurativa. JAMA Dermatol. 2023;159(9):939‐944.37494055 10.1001/jamadermatol.2023.2227 PMC 10372757 · doi ↗ · pubmed ↗
- 8Xiao XM , Yang WZ , Lin LH , Li CR . Two novel nicastrin mutations in Chinese families with acne inversa. J Dermatol. 2020;47(12):e 449‐e 451.32940366 10.1111/1346-8138.15575 · doi ↗ · pubmed ↗