BAPoma, ein seltener Naevus als Schlüssel zur Diagnose des BAP1‐assoziierten Tumorprädisposition‐Syndroms
Lara Racz, Sandra Pasternack‐Ziach, Isabel Spier, Stephan Forchhammer, Claudia Rehkämper, Peter Kind, Julia Reifenberger, Silke Redler

Abstract
Click any figure to enlarge with its caption.
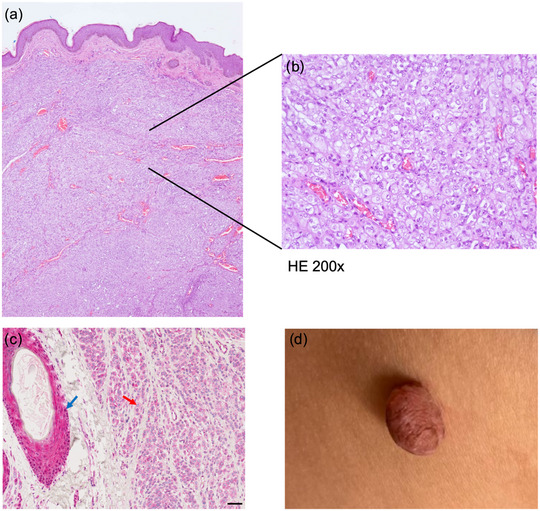 Figure 1
Figure 1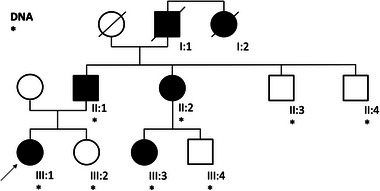 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOccupational and environmental lung diseases · Oral Health Pathology and Treatment · Interstitial Lung Diseases and Idiopathic Pulmonary Fibrosis
Sehr geehrte Herausgeber,
Eine 23‐jährige Frau stellte sich aufgrund größenprogredienter Nävi in unserem Klinikum vor. Es wurde ein Hautkrebsscreening mit body mapping durchgeführt. Aus dem Bereich der dorsalen beziehungsweise ventralen Flanke wurden drei suspekte Nävi exzidiert. Bei zwei dieser drei suspekten Hautveränderungen handelte es sich um junktionale Nävi. Die histologische Untersuchung des Gewebes aus der ventralen Flanke ergab jedoch einen melanozytären Naevus mit partieller BAP1‐Inaktivierung (BAPoma) (Abbildung 1). Immunhistochemisch zeigte sich ein Verlust der nukleären BAP1‐Expression (Abbildung 1). Aus peripheren Blutleukozyten wurde DNS extrahiert und in Einklang mit dem deutschen Gendiagnostikgesetz eine Sequenzierung sowie ein Deletions‐/Duplikationsscreening des Gens BRCA1‐assoziiertes Protein 1 (BAP1) durchgeführt. Dabei wurde die heterozygote pathogene Keimbahnvariante c.1813G>T;p.(Glu605Ter) festgestellt, die zu einem vorzeitigen Stoppcodon führt. Der Nachweis dieser Variante sicherte die Diagnose eines BAP1‐assoziierten Tumorprädisposition‐Syndroms (BAP1‐TPS) bei der jungen Frau. Bei Aufnahme des Familienstammbaums über drei Generationen ergab sich eine Häufung von Tumorerkrankungen in der väterlichen Familie der Index‐Patientin, die sich teilweise in einem frühen Erwachsenenalter manifestiert hatten (Abbildung 2). Interessanterweise wurden bei der Cousine väterlicherseits (III:3) im Alter von 23 Jahren zwei suspekte Hautläsionen exzidiert, die sich beide als BAPome herausstellten. Beim Vater (II:1) wurde im Alter von 47 Jahren die Diagnose eines Urothelkarzinom der Harnblase gestellt, in der Folge trat ein Rezidiv auf. Im Alter von 54 Jahren wurde ein Pleuramesotheliom diagnostiziert. Darüber hinaus hatte sich der Vater der Entfernung zahlreicher Basalzellkarzinome unterzogen. Bei ihm wurde die bei seiner Tochter identifizierte heterozygote pathogene Keimbahnvariante c.1813G>T;p.(Glu605Ter) im BAP1‐Gen ebenfalls festgestellt und damit die Diagnose eines BAP1‐TPS bei ihm gestellt. Bei der Tante väterlicherseits (II:2) wurden seit ihrem 47. Lebensjahr 50 bis 60 adenomatöse Kolonpolypen entfernt, eine pathogene Keimbahnvariante in bekannten Polyposis‐assoziierten Suszeptibilitätsgenen konnte bei ihr nicht nachgewiesen werden. Zwischen dem 40. und 45. Lebensjahr wurden bei ihr mindestens drei Basalzellkarzinome im Halsbereich entfernt. Der Großvater väterlicherseits ist im Alter von 75 Jahren an einem Nierenzellkarzinom gestorben, dessen Schwester verstarb im Alter von 41 Jahren an metastasierendem Brustkrebs. Eine Segregation mit der familiären BAP1‐Variante war bei den verstorbenen Angehörigen nicht mehr möglich. Mit Ausnahme eines Cousins väterlicherseits (III:4) trugen jedoch alle sonst getesteten Familienmitglieder die familiäre BAP1‐Variante (Abbildung 2).
Das BAP1‐assoziierte Tumorprädisposition‐Syndrom wurde erstmalig 2011 beschrieben.1 Dieses seltene familiäre Krebssyndrom wird autosomal‐dominant vererbt und beruht auf pathogenen BAP1‐Keimbahnvarianten, dessen Hauptfunktion die Tumorsuppression auf molekularer Ebene ist. BAP1 wurde aufgrund seiner Interaktion mit dem Gen BRCA1 (BReast CAncer Gene 1) benannt, das durch die Reparatur von DNA‐Doppelstrangbrüchen genomische Stabilität gewährleistet.2 BAP1 wird an Stellen von Doppelstrangbrüchen rekrutiert und unterstützt deren Reparatur durch homologe Rekombination, indem es die Anlagerung von BRCA1 erleichtert.3 Darüber hinaus ist es Bestandteil des Polycomb‐Repressoren‐Deubiquitinase‐Komplexes, der Ubiquitin von Histonen (H2AK119ub) entfernt und dadurch die Transkription reguliert. Ein Verlust von BAP1 stört diesen Prozess, was zu einer veränderten Expression von Genen führt, die in der Zellzykluskontrolle, DNA‐Reparatur und im Zellmetabolismus eine Rolle spielen.4 BAP1‐TPS prädisponiert für verschiedene Tumorentitäten, gutartige melanozytäre Hauttumoren sowie zahlreiche bösartige Tumoren in verschiedenen Organsystemen.5 Wie bei vielen seltenen TPS ist unklar, welche Krebsarten eindeutig mit dem BAP1‐TPS assoziiert sind. Es besteht Konsens, dass der Kernphänotyp des BAP1‐TPS das BAPom, das kutane Melanom, das Aderhautmelanom, das Basalzellkarzinom, das maligne Mesotheliom der Pleura und des Peritoneums sowie das Nierenzellkarzinom umfasst. Darüber hinaus scheint es möglich, dass zu dem Spektrum von BAP1‐TPS auch Meningeome, Cholangiokarzinome, Brust‐, Harnblasen‐ und Lungenkrebs zählen.1, 6, 7 Das BAPom ist häufig die Erstmanifestation dieses seltenen TPS.
Klinisch erscheinen BAP1‐inaktivierte Nävi als kuppelförmige, hautfarbene oder rötliche Papeln. Histologisch stellen sie sich typischerweise als dermale melanozytäre Proliferationen dar, die mit prominenten epitheloiden Zellen kombiniert sind. Letztere können einen nekrotischen Pleomorphismus und reichlich amphophiles Zytoplasma aufweisen und zeigen immunhistochemisch einen Verlust der nukleären BAP1‐Expression.8, 9 Dermatologen sind ideal positioniert, um spezifische klinische Merkmale zu erkennen und eine Keimbahntestung und genetische Beratung zu veranlassen. Dies ist von entscheidender Bedeutung für die Identifizierung von Hochrisikopersonen und deren Einschluss in Screening‐Programme, die darauf abzielen, bösartige Tumoren in einem frühen Entwicklungsstadium zu erkennen und Krebserkrankungen „abzufangen“, bevor sie aggressiv werden. Im Jahr 2023 veröffentlichte ein europäisches Expertengremium detaillierte Empfehlungen zum Umgang mit dem BAP1‐TPS, in denen die Notwendigkeit regelmäßiger Haut‐ und Augenscreenings sowie Bildgebungen der Nieren hervorgehoben wurde.10 Das im vorliegenden Bericht beschriebene Tumorspektrum erweitert die phänotypische Variabilität von BAP1‐TPS. Insbesondere ist dies der erste BAP1‐TPS‐Bericht, in dem kolorektale adenomatöse Polypen (II:2) beschrieben werden, obwohl wir die Möglichkeit nicht ausschließen können, dass sich die kolorektalen Polypen unabhängig von der familiären BAP1‐Variante entwickelt haben.
Der vorliegende Fall zeigt die Schwierigkeiten, die mit der Charakterisierung seltener Tumorsyndrome verbunden sind, für die nur begrenzte wissenschaftliche Erkenntnisse vorliegen. Die Charakterisierung des assoziierten Tumorspektrums, die Ermittlung entsprechender Genotyp‐Phänotyp‐Korrelationen und die Erforschung des Nutzens von Überwachung und klinischem Management sind entscheidende Schritte nach vorn. Zusammenfassend lässt sich sagen, dass unser Bericht eine BAP1‐TPS‐Familie mit kutanen und nichtkutanen Tumormanifestationen beschreibt. BAP1‐TPS ist selten, und aufgrund seines vielfältigen Phänotyps – auch innerhalb von Familien – kann die Diagnose eines BAPoms ein wertvolles Instrument für die Identifizierung dieses Tumorsyndroms darstellen. Die frühzeitige Erkennung von BAP1‐TPS erleichtert die Einleitung von Früherkennungsmaßnahmen, um Malignome zu verhindern, die bei Diagnosestellung weit fortgeschritten sind.
DANKSAGUNG
Wir danken der Familie für ihr Einverständnis, ihren Fall veröffentlichen und histologische/klinische Bilder zeigen zu dürfen.
Open access Veröffentlichung ermöglicht und organisiert durch Projekt DEAL.
INTERESSENKONFLIKT
Keiner.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Testa JR , Cheung M , Pei J , et al. Germline BAP 1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43(10):1022‐1025.21874000 10.1038/ng.912PMC 3184199 · doi ↗ · pubmed ↗
- 2Jensen DE , Proctor M , Marquis ST , et al. BAP 1: a novel ubiquitin hydrolase which binds to the BRCA 1 RING finger and enhances BRCA 1‐mediated cell growth suppression. Oncogene. 1998;16(9):1097‐1112.9528852 10.1038/sj.onc.1201861 · doi ↗ · pubmed ↗
- 3Yu H , Pak H , Hammond‐Martel I , et al. Tumor suppressor and deubiquitinase BAP 1 promotes DNA double‐strand break repair. Proc Natl Acad Sci U S A. 2014;111(1):285‐290.24347639 10.1073/pnas.1309085110 PMC 3890818 · doi ↗ · pubmed ↗
- 4Ge W , Yu C , Li J , et al. Basis of the H 2AK 119 specificity of the Polycomb repressive deubiquitinase. Nature. 2023;616(7955):176‐182.36991118 10.1038/s 41586-023-05841-y · doi ↗ · pubmed ↗
- 5Wiesner T , Obenauf AC , Murali R , et al. Germline mutations in BAP 1 predispose to melanocytic tumors. Nat Genet. 2011;43(10):1018‐1021.21874003 10.1038/ng.910PMC 3328403 · doi ↗ · pubmed ↗
- 6Carbone M , Yang H , Pass HI et al. BAP 1 and Cancer. Nat Rev Cancer. 2013;13(3):153‐159.23550303 10.1038/nrc 3459 PMC 3792854 · doi ↗ · pubmed ↗
- 7Abdel‐Rahman MH , Pilarski R , Cebulla CM , et al. Germline BAP 1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48(12):10.1136/jmedgenet-2011-100156.PMC 382509921941004 · doi ↗ · pubmed ↗
- 8Haugh AM , Njauw CN , Bubley JA , et al. Genotypic and Phenotypic Features of BAP 1 Cancer Syndrome. JAMA Dermatol. 2017;153(10):999‐1006.28793149 10.1001/jamadermatol.2017.2330 PMC 5710339 · doi ↗ · pubmed ↗