Assembly and comparative analysis of four complete mitochondrial genomes of Pulsatilla species
Yanping Xing, Che Bian, Jie Wu, Hefei Xue, Wenxiao Men, Wenjuan Hou, Yutong Huang, Yanchang Huang, Han Zheng, Jianhua Wang, Tingguo Kang, Yanyun Yang, Liang Xu

TL;DR
This study assembles and compares the mitochondrial genomes of four Pulsatilla species to better understand their genetic relationships and medicinal potential.
Contribution
The study provides the first complete mitochondrial genome assemblies for four Pulsatilla species and reveals insights into their genetic variability.
Findings
The four Pulsatilla species have varying mitochondrial genome sizes and gene compositions.
Significant differences in repeat sequences and RNA editing sites were observed among the species.
Phylogenetic analysis grouped the four species into a common subclade, suggesting close genetic relationships.
Abstract
Pulsatilla species, which belong to the Ranunculaceae family, have garnered significant attention due to their remarkable medicinal attributes and ornamental value. In the present study, four mitochondrial genomes (mitogenomes) of Pulsatilla species were assembled and analyzed. The aim was to lay a research foundation for unraveling the genetic interrelationships among these species and the identification of Traditional Chinese Medicine from Pulsatilla species. The mitogenomes of P. chinensis, P. chinensis var. kissii, P. cernua, and P. dahurica were assembled into single circular DNA molecules, with lengths of 878,988 bp, 684,203 bp, 747,621 bp, and 824,625 bp, encoding 53, 40, 40, and 49 protein-coding genes, 13, 14, 20, and 33 transfer RNA genes, and 3, 3, 4, and 3 ribosomal RNA genes, respectively. Repeat sequence analysis found a large number of simple sequence repeats (SSRs) and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
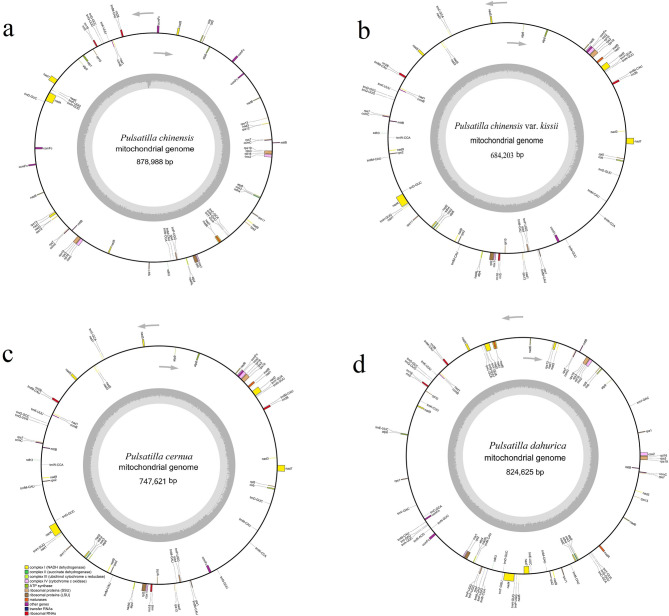 Figure 1
Figure 1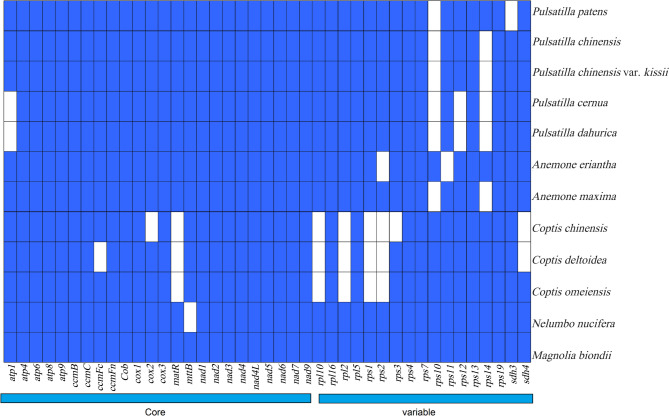 Figure 2
Figure 2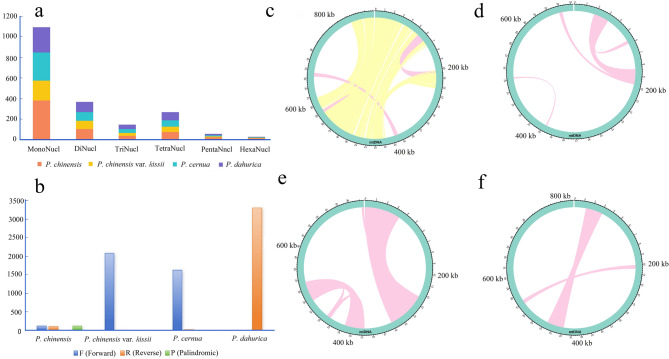 Figure 3
Figure 3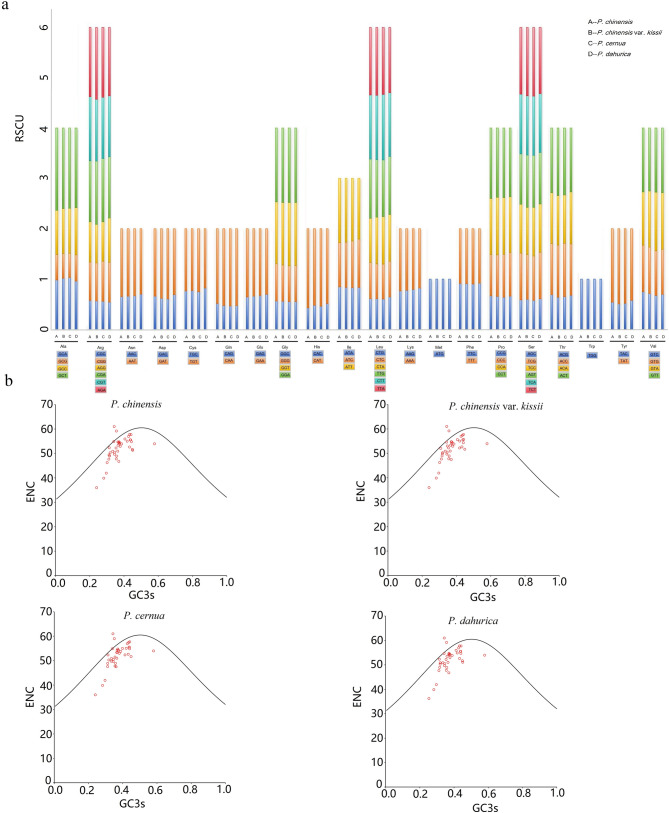 Figure 4
Figure 4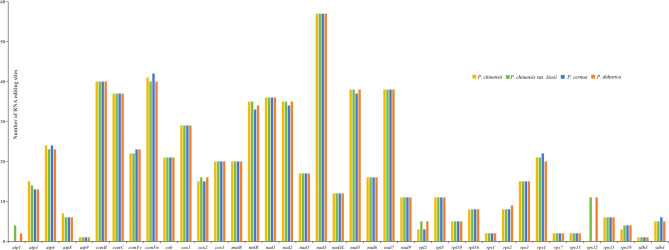 Figure 5
Figure 5 Figure 6
Figure 6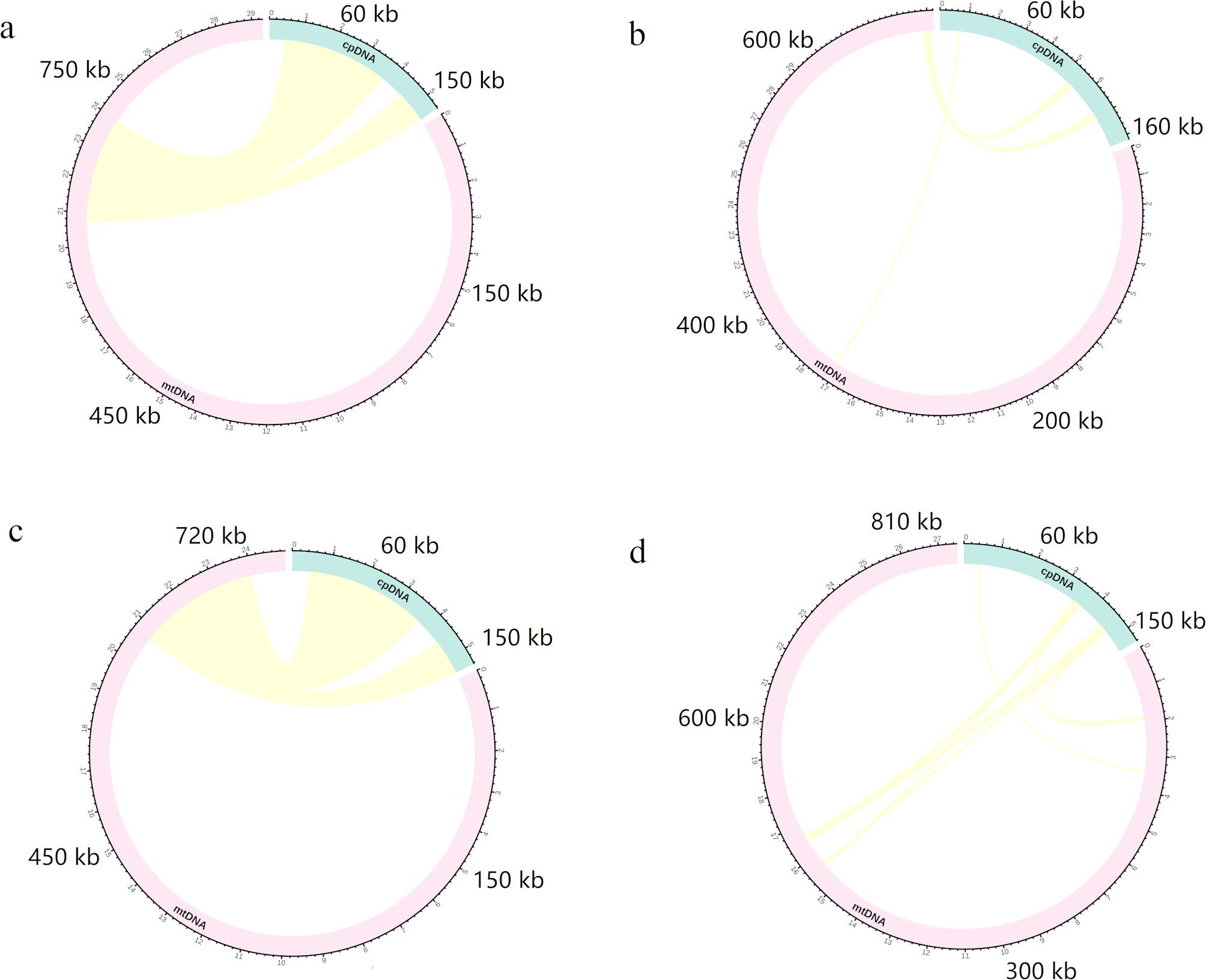 Figure 7
Figure 7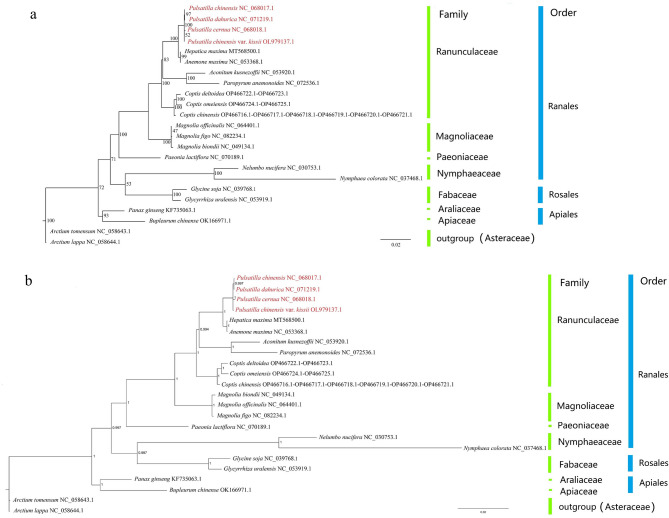 Figure 8
Figure 8 Figure 9
Figure 9- —Liaoning Province Joint Fund
- —Liaoning Provincial Department of Education Basic Research Project Reserve Project for Universities
- —National Natural Science Foundation of China
- —Liaoning BaiQianWan Talents Program
- —Liaoning Provincial Department of Education General Project
- —Key project at central government level: The ability establishment of sustainable use for valuable Chinese medicine resources
- —Research and Demonstration of Key Technologies for High-quality Preparation of Bioactive Ingredients from Plants with Special Efficacy, Sub-project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Microbial Natural Products and Biosynthesis · Fungal Biology and Applications
Introduction
The genus Pulsatilla (Ranunculaceae) comprises approximately 38 species across Europe and Asia, with 11 species found in China [1, 2]. Pulsatilla plants called Pasque-flower have horticultural and medical importance [3, 4]. Pulsatilla chinensis (Bge.) Regel is a commonly used medicinal plant in China. Its roots, known as Pulsatilla radix in traditional Chinese medicine, have been included in the Chinese Pharmacopeia and have been utilized for over 2,000 years to clear away heat and toxins, cool blood, and treat dysentery [5]. Recent pharmacological research showed that its root extracts and polysaccharides exhibit therapeutic effects against cancer and leukemia [6–8]. However, roots of P. chinensis var. kissii (Mandl) S. H. Li & Y. H. Huang, P. dahurica (Fisch. ex DC.) Spreng. and P. cernua (Thunb.) Bercht. & J. Presl. are often misidentified as Pulsatilla radix due to morphological similarity and traditional use. Despite their medicinal properties, these species are considered substitutes. Research has demonstrated chemical differences among P. chinensis, P. cernua, and P. dahurica [9], and only P. chinensis and P. chinensis var. kissii contain anemoside B4 at levels meeting the standards of the Chinese Pharmacopoeia —variations that may impact therapeutic efficacy and safety [5, 10]. Accurate species identification is therefore essential for the proper use of these medicinal plants. P. chinensis var. kissii grows on dry slopes in Liaoning, China. Its basionym, Pulsatilla kissii Mandl (1922), has been suggested as a hybrid between P. chinensis and P. cernua [2], though no definitive evidence supports this claim, although there are reports of hybridization in Pulsatilla species [11]. Chloroplast gene analyses suggest a close relationship between Pulsatilla and P. chinensis var. kissii [12, 13], and molecular identification utilizing ITS2 has proven useful for species discrimination in some Pulsatilla species [14]. Other DNA barcodes (rbcL, trnH-psbA, matK, and ITS) have also been used in phylogenetic studies, though results have been somewhat inconsistent [15]. Expanding the genomic dataset may improve species discrimination and clarify phylogenetic relationships, thus aiding in the development and application of Pulsatilla medicinal resources.
The mitochondrion is an organelle derived from prokaryotic endosymbiont ancestors, and it plays an integral role in cell growth and cell development, impacting overall plant growth and development [16, 17]. For the most part, in seed plants, the mitochondrial genes are inherited maternally. The mitogenome demonstrates a high level of conservation and can function as a foundation for species classification, thereby resolving the issue of distinguishing between closely related species [18]. However, with the increasing reports on plant mitogenomes, numerous studies have revealed that shows notable variability in size, structure, and repeat content even among closely related species, offering valuable insights into plant evolution [19–22]. Meanwhile, mitochondrial genes are associated with plants’ environmental adaptability, tissue growth specificity, and fertility [15, 23–25]. They can also be used to identify the original animal and plant sources of Chinese medicinal materials, showing potential for the development of DNA molecular markers [24, 26, 27]. Therefore, research on mitogenomes can provide new genomic resources for studies on the phylogenetic relationships, evolution, and medicinal material identification of Pulsatilla plants.
This study presents four complete mitogenomes of the Pulsatilla species (P. chinensis, P. chinensis var. kissii, P. cernua, and P. dahurica) (Fig. S1) for the first time. The genome structure, gene composition, codon usage, RNA editing sites, and repeat sequences were investigated and analyzed. A phylogenetic analysis of 23 mitogenomes was undertaken based on 14 protein-coding gene (PCG) sequences. The goals were to clarify the characteristics of the mitogenomes of P. chinensis, P. chinensis var. kissii, P. cernua, and P. dahurica, as well as the differences among them. And by mitochondrial genes, the phylogenetic relationships and evolutionary positions of these species and variety will be explored. By analyzing the mitogenomes of these species, it can provide a research foundation for the environmental adaptation mechanisms of plants in the Pulsatilla genus and the improvement of cultivated varieties, while also offering basic data for developing molecular markers used in the identification of traditional Chinese medicines.
Results
General features of the sequenced mitogenomes of the four mitogenomes
The complete mitogenomes of P. chinensis, P. chinensis var. kissii, P. cernua, and P. dahurica were assembled as circular DNA molecules with lengths of 878,988 bp, 684,203 bp, 747,621 bp, and 824,625 bp, respectively and GC contents of 46.86%, 46.35%, 45.4%, and 46.46% (Fig. 1; Table 1). The average read coverage depth of the 4 samples ranged from 327.72X to 1243.97 (Fig. S2). All four mitogenomes were resolved to have a complete single circular molecular structure (Fig. S3). These genome sizes show considerable variation, differing notably from those of other Ranunculaceae species such as Anemone maxima and Coptis species [20, 28]. The number of PCGs identified in each mitogenome was 53 (P. chinensis), 40 (P. chinensis var. kissii), 40 (P. cernua), and 49 (P. dahurica) (Table S1). The genes atp1 and rps12 were present in P. chinensis and P. chinensis var. kissii but absent in P. dahurica and P. cernua. According to the reported classification scheme [29], mitochondrial PCGs can be categorized into “core” and “variable” genes. The “variable” gene rps10 was absent across all five Pulsatilla species (Fig. 2), highlighting the variability of gene content even among closely related taxa.
For all four mitogenomes, there were nine PCGs contained introns: ccmFc, cox2, rpl2, and rps3 each harbored one intro; nad1, nad2, nad5, and nad7 each contained four introns; and nad4 contained three introns (Table S2). The number of tRNA genes was 13 in P. chinensis, 14 in P. chinensis var. kissii, 20 in P. cernua, and 33 in P. dahurica. Correspondingly, 3, 3, 4, and 3 rRNA genes were identified. Additionally, the number of open reading frames (ORFs) was 251 (P. chinensis), 199 (P. chinensis var. kissii), 251 (P. cernua), and 241 (P. dahurica), with total ORF lengths of 119,223 bp, 92,253 bp, 138,696 bp, and 109,428 bp, respectively (Table S3).
Fig. 1. Circular maps of the P. chinensis (a), P. chinensis var. kissii (b), P. cernua (c), and P. dahurica (d) mitogenomes. Genes depicted on the outer and inner rings are transcribed in the clockwise and counterclockwise directions, respectively. Functional categories were indicated by color-coding
Table 1. Summary of four mitogenomes sequencing statistics of Pulsatilla speciesP. chinensis**P. chinensis var. kissii**P. cernua**P. dahuricaGenome Size (bp)878,988684,203747,621824,625GC Content (%)46.8646.3545.446.46protein-coding genes Number53404049Gene Total Length (bp)47,43635,43335,08542,996tRNA (transfer RNA)13142033rRNA (ribosome RNA)3343Gene Average Length (bp)895886877877Gene’s GC Content (%)43.9643.8343.6544.17% of Genome (Genes)5.45.184.695.21Intergenic region Length (bp)831,552648,770712,536781,629Intergenic’s GC Content (%)47.0346.4945.4946.59% of Genome (Intergenic)94.694.8295.3194.79
Fig. 2. The presence of protein coding genes of mitogenomes in plants of the genus Pulsatilla and its related species, with blue boxes indicating the presence of those genes and white boxes indicating the absence of such genes. Core represented core genes and variable represented variable genes
Repeat sequence analysis
Six types simple sequence repeats (SSRs) were identified in the mitogenomes of Pulsatilla species, with 631 in P. chinensis, 369 in P. chinensis var. kissii, 474 in P. cernua, and 481 in P. dahurica (Fig. 3a). Notably, P. chinensis uniquely harbored 41 SSRs exceeding 20 bp in length (Table S4). Across all four mitogenomes, monomeric SSRs were the most abundant, followed by dimeric SSRs and etrameric SSRs. The proportion of A/T in all SSRs was the highest, with 30.74%, 45.80%, 51.69%, and 44.28% in P. chinensis, P. chinensis var. kissii, P. cernua, and P. dahurica, respectively.
Long repeat sequences (LRSs) were also abundant in all four mitogenomes, categorized into forward (F), reverse (R), and palindromic (P) types (Fig. 3b). P. dahurica exhibited the highest number of LRSs (3,299 pairs), all of which were R-type. In contrast, P. chinensis (349 pairs) contained all three types. The length of LRSs ranges from 200 to 26,008 bp in P. chinensis, 30–21,799 bp in P. chinensis var. kissii, 30–71,126 bp in P. cernua, and 30–19,430 bp in P. dahurica (Table S5). P. chinensis contained 43 LRSs exceeding 1 kb, including eight over 10 kb and three over 20 kb. The other three species had significantly fewer LRSs over 1 kb. Notably, P. cernua had only six LRSs above 1 kb, yet included one over 70 kb and another over 30 kb (Fig. 3c-f).
Fig. 3. Repeats were detected in the mitogenomes of P. chinensis, P. chinensis var. kissii, P. cernua, and P. dahurica. a, Type and number of detected SSRs; b, Type and number of long repeat sequences. Repetitive sequences of more than 1 kb in the four mitogenomes: c, P. chinensis; d, P. chinensis var. kissii; e, P. cernua; f, P. dahurica; the yellow lines represented P (Palindromic), and the pink lines represented F (Forward) LRSs, respectively
Codon bias ratios analysis
The relative synonymous codon usage (RSCU) values were assessed for mitochondrial PCGs in the four Pulsatilla mitogenomes (Fig. 4a). Each mitogenomes contained exhibited 29 codons with RSCU values greater than 1, indicating codon usage bias. A strong preference was observed for A and T at the third codon position, while the first base showed a more balanced distribution. Among all codons, GCT (Ala) had the highest RSCU value (1.635). Overall, codon usage patterns were consistent across the four species, with minor exceptions: GCA (Ala) was more frequently used in P. chinensis var. kissii and P. cernua, while ACA (Thr) was favored in P. chinensis and P. dahurica. The start codon AUG (Met) and UGG (Trp) both maintained RSCU values of 1, aligning with patterns reported in other species [30, 31]. The effective number of codons (ENCs) revealed similar codon usage trends among the species (Fig. 4b and Table S6). Most PCGs clustered along the expected curve for 24–58% GC3. There were two PCGs, nad9 and sdh3, whose ENC values were above the standard curve. A majority of the points lay below the standard curve, especially those of rps13 and atp9, which were far below the standard curve and whose ENC values were less than 40.
Fig. 4. Codon bias and ENC plot of the PCGs in four mitogenomes of Pulsatilla species. a, RSCU values of the four mitogenomes; b, ENC plot of the four mitogenomes. Standard curve line was calculated as follows: ENC = 2 + GC3 + 29/(GC3s^2^ + (1-GC3)^2^)
RNA editing sites analysis
RNA editing refers to the addition, deletion, or replacement of nucleotides in RNA, causing changes in genetic information as the nucleotide sequences are different from corresponding genomic DNA sequences. This process occurs widely in organisms [32]. In the four Pulsatilla mitogenomes analyzed, RNA editing patterns in PCGs were largely consistent. A total of 691, 694, 679, and 677 RNA editing sites were identified in P. chinensis, P. chinensis var. kissii, P. cernua, and P. dahurica, respectively (Fig. 5). The number of editing sites varied by gene, from just one site in atp9 to 57 in nad4. All editing events were cytosine-to-uracil (C-to-U) conversions. These edits led to the formation of start codons in nad1, cox2, nad5, and atp4, and stop codons in atp6, rpl16, and ccmFC (Table S7).
.
Fig. 5. Distribution of RNA editing sites across the mitochondrial protein - coding genes of the four investigated plants
Collinearity and plastid-derived region analysis
Sequence similarity analysis revealed extensive collinear blocks among the four Pulsatilla mitogenomes (Fig. 6). The collinear regions accounted for 65.0% (568 kb) and 91.7% (756 kb) of the total mitogenome length in P. chinensis and P. dahurica, respectively, with a total of 89 collinear blocks identified, including 24 blocks larger than 10 kb and 57 blocks between 1 and 10 kb in length. The collinear sequences accounted for 83.0% (569 kb) and 85.0% (744 kb) of the total sequences of P. chinensis var. kissii and P. chinensis, respectively, with a total of 100 collinear blocks identified, including 26 blocks larger than 10 kb and 67 blocks between 1 and 10 kb in length. Between P. chinensis var. kissii and P. cernua, there were 72 collinear blocks, with 23 longer than 10 kb and 46 longer than 1 kb. The collinear sequences accounted for 72.0% (495 kb) and 78.0% (582 kb) of the total sequences of P. chinensis var. kissii and P. cernua, respectively. The collinear regions accounted for 81.0% (603 kb) and 62.0% (607 kb) of the total mitogenome length in P. patens and P. cernua, respectively. Synteny analysis indicated a high degree of synteny conservation among the mitogenomes of species within the genus Pulsatilla. Gene-level collinearity showed more divergence than whole-genome comparisons (Fig. S4, Table S9). Between P. chinensis var. kissii and P. chinensis, 50 collinear blocks (PCGs) accounted for 82% and 62% of their total gene sequences, respectively. In contrast, the comparison between P. chinensis var. kissii and P. cernua revealed 36 collinear blocks (PCGs), comprising 74% and only 17% of the total gene sequences, respectively. The comparison between P. chinensis and P. dahurica identified just 18 collinear gene fragments (PCGs), each representing only 4% of their respective total gene sequence lengths.
Fig. 6. Multiple synteny plot of the five mitogenomes. Homologous sequences of more than 500 bp between two species were connected by arcs. Red arcs represented inverted sequences and gray arcs represented forward sequences
A comparative analysis of mitochondrial plastid sequences (MTPTs) among the four Pulsatilla species (Fig. 7) revealed that MTPT content and distribution varied notably: P. cernua contained 97 kb across 2 homologous fragments, P. chinensis had 19 kb (7 fragments), P. dahurica 13 kb (26 fragments), and P. chinensis var. kissii 7 kb (3 fragments). These MTPTs accounted for 13.1%, 2.0%, 1.6%, and 1.1% of the total mitogenome lengths, respectively.
Fig. 7. Plastid-derived region of mitochondrial sequences of the Pulsatilla species: a, P. chinensis; b, P. chinensis var. kissii; c, P. cernua; d, P. dahurica
Phylogenetic analysis
Phylogenetic trees constructed using maximum likelihood (ML) and Bayesian inference (BI) were largely congruent, with most families and orders forming monophyletic groups (Fig. 8). Within the Ranunculaceae family, Pulsatilla species formed a well-supported monophyletic clade. ML analysis grouped P. chinensis and P. dahurica together, while P. chinensis var. kissii clustered with P. cernua, though with weak support (52%) (Fig. 8a). In contrast, BI analysis grouped P. chinensis and P. cernua, with P. chinensis var. kissii and P. dahurica forming separate branches (Fig. 8b). Overall, Pulsatilla showed closer genetic affinity to Anemone and Hepatica. Gene concordance factor (gCF) and site concordance factor (sCF) analyses of the phylogenetic tree showed that this topological structure was not well-supported (Fig. S5). Our analysis of the shared protein sequences from four Pulsatilla species (used for constructing the phylogenetic tree in this study) revealed that these sequences were almost identical, which may be the reason why this phylogenetic tree cannot clarify the evolutionary relationships among species within Pulsatilla. Furthermore, we reconstructed ML and BI phylogenetic trees for Hepatica maxima and 4 Pulsatilla species using 21 conserved mitochondrial proteins. The results showed that the two phylogenetic trees exhibited different topological structures with low support values (Fig. S6). In contrast, the phylogenetic tree constructed using shared proteins from the chloroplast genome was consistent with previous research results (Fig. 9) [12, 13].
Fig. 8. Phylogenetic trees constructed using 14 PCGs of 23 mitogenomes. a, Maximum likelihood (ML) tree; b, Bayesian inference (BI) tree
Fig. 9. Phylogenetic trees constructed using 50 PCGs of 24 chloroplast genomes.** a**, Maximum likelihood (ML) tree; b, Bayesian inference (BI) tree
Discussion
Plant mitogenomes are highly dynamic, with substantial variation in size, structure, and gene content—an ongoing focus of evolutionary research [33, 34]. In this study, the complete mitogenomes of P. chinensis, P. chinensis var. kissii, P. cernua, and P. dahurica were assembled. All four exhibited circular configurations, consistent with other species such as Camellia duntsa, Hepatica maxima,* Bupleurum chinense*, and Avena longiglumis [21, 28, 31, 35], but differing from P. patens, which has three linear mitochondrial chromosomes [1]. Such structural variation within genera has also been reported in Rhodiola and Calla [25, 36]. The mitogenome sizes of the four Pulsatilla species ranged from 684,203 bp to 878,988 bp—a nearly 100 kb difference—while that of P. patens extends to 986,613 bp [1], illustrating the size variation within the genus. The entire mitogenomes of four Pulsatilla species have a GC content ranging from 45.4% to 46.86%, which is relatively conserved compared with their genome sizes. Deletions of protein-coding genes (PCGs) are common in plant mitogenomes [29, 37], and Pulsatilla species contain between 40 and 53 PCGs, with gene number generally correlating with genome size [29]. The results demonstrated that compared with core genes, variable genes in the mitogenome are more prone to loss [38]. Consistent with earlier findings, the rps10, rps14 gene is missing from all four Pulsatilla mitogenomes [1, 34] Notably, the core gene atp1 and the variable gene rps12 are absent in P. dahurica and P. cernua. These findings showed that the PCGs of mitogenomes among Pulsatilla exhibited diversity, potentially serving as the scientific references for species identification.
Plant mitogenomes are rich in repetitive sequences, which may contribute to their structural diversity and complexity [16, 39]. The Pulsatilla species analyzed in this study contained numerous repeats, including SSRs and LRSs. Each species harbored over 300 SSRs, with P. chinensis exhibiting the highest number, exceeding 600. Furthermore, in addition to significant differences in quantity (ranging from 369 to 631), the types of SSRs also showed variations (Table S4), and this phenomenon has also been reported in other species [38]. Multiple LRSs longer than 10 kb were identified, with significant variation in the number of LRSs >1 kb among species. P. chinensis had the highest LRS abundance, while P. cernua contained the longest LRS at 70 kb. LRSs are widespread within Pulsatilla, which may indicate extensive insertion and recombination events during the evolution of mitogenomes [24]. Such variability in repetitive sequence patterns may underlie the observed differences in mitogenome size and PCG content across Pulsatilla species [36].
The RSCU values reflects relationship between observed and expected codon frequencies, allowing assessment of species-specific codon preferences and evolutionary patterns [40]. In all four Pulsatilla mitogenomes, 29 codons showed RSCU values >1, most of which ended in A or T bases. This consistent codon preference aligns with patterns observed in other plant genera [35] and varies minimally among the Pulsatilla species. Similarly, ENC values were comparable across species [41, 42], indicating conserved codon usage bias within the genus. As in previous studies, most points fell below the expected curve in ENC-GC3s plots, suggesting that codon bias is primarily driven by natural selection or other non-neutral factors [43]. Notably, Atp9 and rps13 deviated substantially below the curve, reflecting stronger codon bias and higher mutational susceptibility relative to other genes [44].
RNA editing, a post-transcriptional modification present in all higher plants [32], helps maintain amino acid sequence conservation in essential mitochondrial proteins [45]. Our analysis showed that P. chinensis and P. chinensis var. kissii had more RNA editing sites in PCGs than P. cernua and P. dahurica. Each of the four mitogenomes contained over 600 editing sites (677–694), fewer than the 900 sites found in P. patens [1]. The distribution and pattern of RNA editing vary markedly among different losely related lineages [38]. C-to-U (C to T) editing dominated, consistent with other plant mitogenomes [1, 36]. RNA editing occurred in nearly all PCGs of Pulsatilla, likely playing a significant role in mitochondrial function [46]. These sites offer valuable targets for studying metabolic regulation of active compounds and stress responses in Pulsatilla.
Collinearity analysis, which examines homologous genes and sequence alignments, facilitates the study of evolutionary relationships among species [30]. The mitogenomes of the five Pulsatilla species exhibited extensive collinear blocks, reflecting high homology. Their evolution involved frequent sequence breaks and fusions [36]. Collinear regions accounted for 65% and 91.7% of the total mitogenome lengths in P. chinensis and P. dahurica, respectively, with the higher proportion in P. dahurica likely due to its abundance of LRSs, indicating that species with closer genetic relationships always share the majority of sequences [47]. Relatively, the existence of regions without homology highlights their uniqueness in this specific mitogenome, which holds important implications for subsequent studies related to genetics, growth, and development.
Gene transfer from plastids to mitochondria is a common evolutionary process in angiosperms [19, 47]. Among the four Pulsatilla mitogenomes, P. cernua contained 97 kb of MTPTs, representing 13.1% of its total mitogenome length—substantially higher than in the other three species. Although this exceeds the typical 1–10% range of plastid-derived regions in mitogenomes [48], similar proportions have been reported in three Melastoma species, with M. dodecandrum reaching approximately 15.16% [49]. This suggests that gene transfer from plastids to mitochondria may have occurred after P. cernua diverged from the other three species. The MTPTs may also contribute to the variation in Pulsatilla mitogenome size [48]. Additionally, mitogenome sequences can transfer back to plastomes [50, 51], indicating a dynamic, bidirectional gene exchange between mitochondria and chloroplasts rather than strict conservatism, even among closely related species.
Consistent with prior studies, species within the same genus or family clustered together in phylogenetic trees constructed from shared mitochondrial PCGs, reflecting congruence between traditional taxonomy and molecular data [22, 30]. The three Pulsatilla species and one variety formed a distinct clade. These results indicated that the mitogenome data can be effectively applied to distinguishing Pulsatilla species from species of other genera. Currently, mitogenome data, chloroplast genome data, and nuclear genome data are widely used in studies on species evolutionary relationships, but the results of these studies are often incongruent [4, 15]. Such phylogenetic incongruence among is well documented in evolutionary studies [38, 52–56] and may arise from processes including organelle capture (introgression), organelle genome recombination, and incomplete lineage sorting of ancient polymorphic organelle genomes [54]. Additionally, hybridization-driven independent organelle replacement can cause mitochondria and chloroplasts to carry differing parental evolutionary signals, leading to discordant phylogenies [15, 54]. For example, cucumber hybrid offspring inherit chloroplast genomes maternally but mitogenomes paternally [57]. However, our research results revealed that the PCGs in the mitogenomes of Pulsatilla plants provide low support for the evolutionary relationships among these four plant species. This may be because their protein sequences exhibit high conservation, such that insufficient variations have accumulated for species delimitation. This high conservation may partly result from the fact that the mutation rate of mitogenomes is generally lower than that of chloroplast genomes in plants [58]. If P. chinensis var. kissii was a hybrid, it also provides a plausible explanation for the species differentiation phenomenon in this genus [57]. In terms of mitogenome size and gene content, P. chinensis and P. dahurica share similarities, while P. chinensis var. kissii clusters with P. cernua. However, synteny analysis revealed that the mitogenome of P. chinensis var. kissii shows higher synteny with P. chinensis than with P. cernua. This discrepancy may be attributed to the relatively high level of MTPS in P. cernua. Although these findings do not conclusively prove a hybrid origin for P. chinensis var. kissii, mitogenomic data offer valuable insights into Pulsatilla phylogeny.
Conclusions
We assembled the complete mitogenomes of P. chinensis, P. chinensis var. kissii, P. cernua, and P. dahurica, analyzing their genome organization, gene content, plastome-to-mitogenome horizontal gene transfer, and phylogenetic relationships. Considerable variation was observed in genome size (ranging from 684,203 bp in P. chinensis var. kissii to 878,988 bp in P. chinensis) and gene number (40 to 53 genes). Notably, P. cernua and P. dahurica lacked the atp1 and rps12 genes. All mitogenomes contained abundant repetitive sequences and homologous DNA fragments, with marked interspecific differences. Codon usage bias and RNA editing site patterns were similar across species. Phylogenetic analysis based on mitochondrial PCGs confirmed the monophyly of Pulsatilla. These results provide valuable genomic resources for plant mitogenome research and offer important insights into the genetic diversity and evolution of Pulsatilla, with potential applications in molecular breeding.
Materials and methods
Sampling, DNA extraction, and sequencing
Fresh leaves of all four Pulsatilla species were collected from the Herb Garden of Dalian Campus, Liaoning University of Traditional Chinese Medicine, Dalian, Liaoning, China (39°06′N, 121°57′E) (Fig. S1). These plants, transplanted from their natural habitats, had been growing for over three years. According to the Regulations of the People^’^s Republic of China on Wild Plants Protection, these species are not classified as nationally key protected wild plants. Article five of the regulations stipulates that the scientific research on wild plants and in situ and ex situ protection of wild plants is encouraged and supported. All operations were carried out in accordance with guidelines in the Specification on Good Agriculture and Collection Practices for Medicinal Plants (GACP; Number: T/CCCMHPIE 2.1–2018). The study was approved by School of Pharmacy, Liaoning University of Traditional Chinese Medicine. The voucher specimens (P. chinensis, 20210503001LY; P. chinensis var. kissii, 20210503002LY; P. cernua, 20210503003LY; P. dahurica, 20210703011LY) were identified by Professor Liang Xu of Liaoning University of Traditional Chinese Medicine and deposited in the Chinese Medicine Specimen Hall of Liaoning University of Traditional Chinese Medicine. Mitochondria were isolated from leaves using density gradient centrifugation and treated with DNase I (Promega, Madison, USA) to remove genomic DNA contamination. Both short-read (Illumina) and long-read (PacBio Sequel II) sequencing technologies were employed. Approximately 2 µg of mitochondrial DNA was used to construct SMRTbell libraries with the PacBio Express Template Prep Kit 2.0. Libraries were multiplexed, size-selected with BluePippin (cutoff: 5000 bp), and sequenced on the PacBio Sequel II platform after annealing and binding via SMRT Link. For Illumina sequencing, approximately 1µg of mitochondrial DNA was sonicated to ~ 500 bp using the Covaris M220 system. The sonicated DNA was purified using a TIANgel Midi Purification Kit, and a sequencing library was constructed using the NEBNext^®^Ultra™ DNA Library Prep Kit for Illumina^®^ (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s instructions. Sequencing was performed on an Illumina NovaSeq 6000 with 150 bp paired-end reads. Short reads were quality checked using FastQC and trimmed with Trimmomatic (ILLUMINACLIP: TruSeq-PE.fa:2:30:10 LEADING:3 TRAILING:3 MINLEN:75). Long reads were base-called using Albacore v2.1.7 (mean_qscore > 7), demultiplexed by barcode, and converted to FASTA with Samtools Fasta (http://www.htslib.org/doc/samtools.html).
Genome assembly and annotation
The mitogenomes were assembled using two complementary strategies. The first involved de novo assembly of short clean reads with GetOrganelle v1.6.4, followed by extraction of mitochondrial contigs through BLAST v2.8.1 + alignment against mitochondrial protein-coding genes from the plant mitogenome database. Potential long mitochondrial reads were identified by mapping PacBio long reads to these contigs using BLASR v5.1 and subsequently assembled with Canu v2.1.1. The second strategy directly assembled all PacBio long reads using Canu. Draft contigs from both approaches were refined by mapping short clean reads using BWA and polished with Pilon v1.22. Circularity of contigs was confirmed using MUMmer 3.23, and the final corrected contigs from both methods were aligned with MUMmer for consistency. The assembly results were visualized and manually refined with Bandage v 0.8.1 software to obtain preliminary results [59]. Coverage depth was determined via Samtools (v 0.9) [60].
Mitochondrial gene annotation was conducted using the online GeSeq tool with default parameters to predict PCGs, transfer RNA (tRNA), and ribosomal RNA (rRNA) genes [61]. PCG locations were verified via BLAST against reference mitochondrial genes from Liriodendron tulipifera (GenBank accession MK340747.1). Start/stop codons and intron/exon boundaries were manually curated in SnapGene Viewer using the reference mitogenome. Mitogenome maps were generated with OGDRAW [62]. Functional annotation of genes involved BLAST searches (E-value ≤ 10⁻⁵) against several protein databases: NCBI Non-redundant (Nr), Swiss-Prot, Clusters of Orthologous Groups (COGs), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Ontology (GO). ORFs were identified using getorf (EMBOSS 6.6.0, http://emboss.sourceforge.net/) with parameters set to -table 1 and -size 300, corresponding to a minimum length of 100 amino acids.
Sequence analysis
SSRs were identified using MISA software with parameters set for unit sizes and minimum repeats as follows: 1–8, 2–5, 3–4, 4–3, 5–3, and 6–3, and a minimum distance of 100 bp between SSRs. LRSs were detected using REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer?id=reputer_manual_manual) with a minimum repeat length of 30 bp, Hamming distance of 3, and a maximum of 5000 repeats (equivalent to 1e-3). Four types of repeats were identified: forward (F), reverse (R), complementary (C), and palindromic (P). Codon usage bias was analyzed by calculating relative synonymous codon usage (RSCU) values using the cusp tool from EMBOSS v6.6.0. The effective number of codons (ENC) for protein-coding genes (PCGs) was evaluated via an online platform (http://cloud.genepioneer.com:9929/#/tool/alltool/detail/290). RNA editing sites were predicted using PREPACT3 (v3.12.0; http://www.prepact.de/) with a cutoff threshold of 0.2.
Plastid-derived region analysis
Plastome sequences of the four species (P. chinensis MK860682, P. chinensis var. kissii MK860683, P. cernuaMK860687, and P. dahurica MK860685) were retrieved from the NCBI database. Homologous sequences between each species’ mitogenome and plastome were identified using BLAST v2.10.1 with default parameters. The results were visualized using the Circos package via an online platform (http://210.22.121.250:35588/CloudPlatform/home).
Collinearity analysis of Pulsatilla species
To investigate intergenomic rearrangements and evolutionary relationships, pairwise mitogenome comparisons of the five Pulsatilla species—including P. patens, whose mitogenome data were downloaded from NCBI (accession numbers: MZ420977, MZ420978, MZ420979)—were conducted using BLASTN (parameters: e-value 1e-5, word size 9, gap open 5, gap extend 2). Collinear blocks were defined as homologous sequences longer than 500 bp. Multiple synteny was visualized with MCScanX [63].
Phylogenetic analysis
Mitogenomes of 17 closely related species and two outgroups (Arctium lappa, NC_058644; A. tomentosum, NC_058643) were selected from NCBI (https://www.ncbi.nlm.nih.gov) based on phylogenetic affinity. The mitogenome of P. patens was excluded due to unavailable annotation files. Using OrthoFinder v2.3.14, 14 conserved protein-coding genes (PCGs) were identified across the 23 mitogenomes: atp9, nad6, nad3, cob, nad4, rpl16, ccmFn, atp8, nad7, atp4, nad2, cox3, ccmC, and ccmB. Sequences were aligned with MUSCLE v3.8.1551 and refined using Gblocks to remove poorly aligned regions and gaps. The filtered alignments were concatenated for phylogenetic reconstruction. Bayesian inference was performed in MrBayes v3.2.6 using two hot and two cold chains for 10 million generations, sampling every 1,000 generations. The first 25% of trees were discarded as burn-in. Maximum likelihood (ML) analysis was conducted using IQ-TREEv1.6.12. The best-fit substitution model (HIVw + G + F) was determined by Modeltest v3.4 based on the Bayesian Information Criterion (BIC). Considering that ML and BI phylogenetic trees have similar topological structures in this study, we performed gene concordance factor (gCF) and site concordance factor (sCF) analyses on the ML phylogenetic tree, and calculated these factors using IQ-TREE v.2.4.0 [64]. Phylogenetic tree based on chloroplast genomes Fifty homologous single-copy protein-coding genes (including accD, atpA, atpB,* atpE*, atpH, atpI, cemA, clpP, matK, ndhA, ndhC, ndhD, ndhE, ndhG, ndhI, ndhJ, petA, petB, petG,* petL*, petN, psaA, psaB, psaC, psaI, psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbJ, psbK, psbT, psbZ, rbcL, rpl20, rpl33, rpl36, rpoA, rpoB, rpoC2, rps2, rps4, rps11, rps14, rps18, ycf3) were selected from 24 samples via OrthoFinder v2.3.14. After individual alignment with MAFFT v7.429, ambiguous regions (gap-containing sites) were trimmed by Gblocks 0.91b, and sequences were concatenated for phylogenetic tree construction. Outgroups: A. lappa NC_058644 and A. tomensum (Download from the Arctium lappa database: http://210.22.121.250:41352/). ML tree: Constructed with IQ-TREE v1.6.1; best model (GTR + F + R3 via BIC) selected by ModelFinder, bootstrap = 1000. Bayesian tree: Built with MrBayes 3.2.6, using 2 hot + 2 cold chains, run for 10,000 generations, sampled every 1000 generations. First 25% trees discarded as burn-in, remaining used for consensus tree.
Supplementary Information
Supplementary Material 1: Fig. S1 The four plants of Pulsatilla species Fig. S2 Sequencing depth and coverage map of the mitogenomes of *P. chinensis *(a), P. chinensis var.kissii (b), P. cernua(c), *P. dahurica *(d) Fig. S3 The assembly graph of mitogenomes displayed in Bandage. (a) Raw assembly networks showing complex reticulation; (b) Curated circular structures inferred from repeat masking and validation Fig. S4 Multiple synteny genes of the four mitogenomes Fig. S5 Maximum likelihood Phylogenetic trees constructed using 14 PCGs of 23 mitogenomes. The tree were annotated with supports which were indicated by concordance factors (UFB/gCF/sCF) Fig. S6 Phylogenetic trees constructed using 21 PCGs (atp4, atp6, atp8, atp9, ccmB, cob, cox1, cox3, nad1, nad2, nad4, nad4L, nad5, nad7, rpl2, rpl5, rpl10, rps4, rps11, sdh3, sdh4) of 5 mitogenomes. (a) Maximum likelihood (ML) tree; (b) Bayesian inference (BI) tree.
Supplementary Material 2: Table S1 Genes predicted in the four mitogenomes of Pulsatilla species Table S2 The introns of protein-coding genes in four mitogenomes Table S3 The ORFs of four mitogenomes of* Pulsatilla* species Table S4 The SSR of four mitogenomes of Pulsatilla species Table S5 The Long repeat sequences of four mitogenomes of Pulsatilla species Table S6 ENC vaule of four mitogenomes of Pulsatilla species Table S7 The RNA-editing sites of four mitogenomes of Pulsatilla species. Table S8 Collinear blocks and their matched genes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lindell T. Breeding systems and crossing experiments in Anemone patens and in the Anemome pulsatilla group (Ranuncdaceae). Nordic journal of botany. Nord J Bot. 1998;18:549–61.