An Unusual Gastrointestinal Presentation of Sjogren's Syndrome: A Case Report
Oudai Sahwan, Fares Jamal, Amani Elshaer, Lin Batha, Tala Shahin, Michael M. Pham

TL;DR
This case report describes a rare instance of Sjogren's syndrome affecting the small intestine, highlighting the diagnostic challenges and treatment response.
Contribution
This is the first reported case of Sjogren's syndrome involving the small intestine, emphasizing the need for autoimmune enteropathy consideration.
Findings
The patient exhibited cyclic gastrointestinal symptoms linked to Sjogren's syndrome, not typically associated with bowel involvement.
Treatment with Rituximab led to clinical improvement, suggesting autoimmune enteropathy as a possible mechanism.
Standard testing failed to detect autoimmune processes, underscoring limitations in current diagnostic approaches.
Abstract
Sjögren's syndrome (SS) is a chronic autoimmune disorder characterized by lymphocytic infiltration and destruction of exocrine glands, commonly involving the salivary and lacrimal glands, leading to dry mouth and eyes. While SS primarily affects the upper gastrointestinal tract, this case presents a rare small intestine involvement. This patient was diagnosed with SS in 2018 with positive antinuclear antibodies (ANA), rheumatoid factor (RF), SSA antibodies, and salivary lip gland biopsy. By 2021, she developed unusual cyclic episodes of nausea, vomiting, and poor oral intake, resulting in significant weight loss and recurrent hospitalizations. She had frequent hospitalizations due to her recurrent symptoms. Evaluations included esophagogastroduodenoscopy (EGD) and colonoscopy, but pathology was nondiagnostic. Several abdominal and pelvic computed tomography (CT) scans demonstrated…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
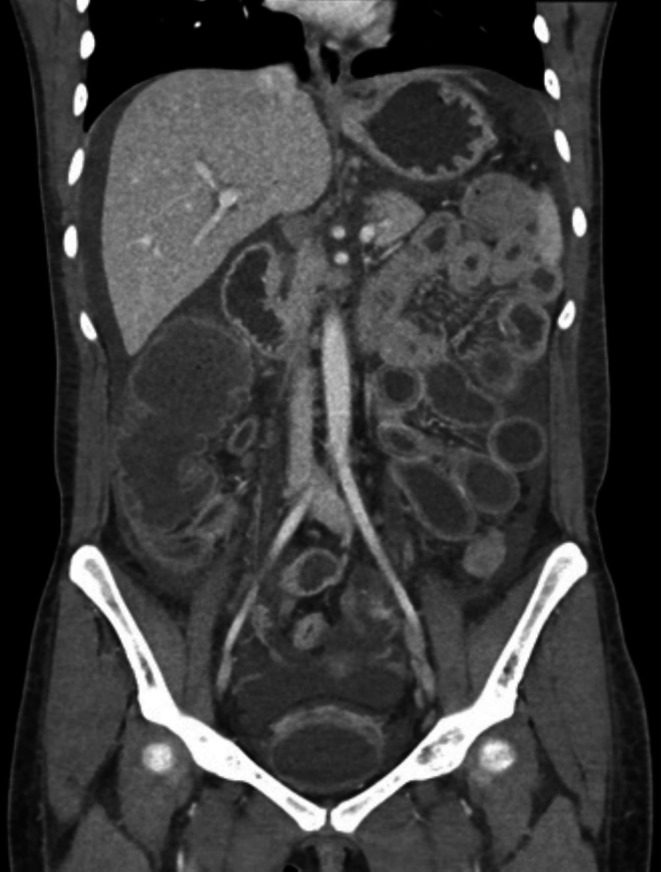 FIGURE 1
FIGURE 1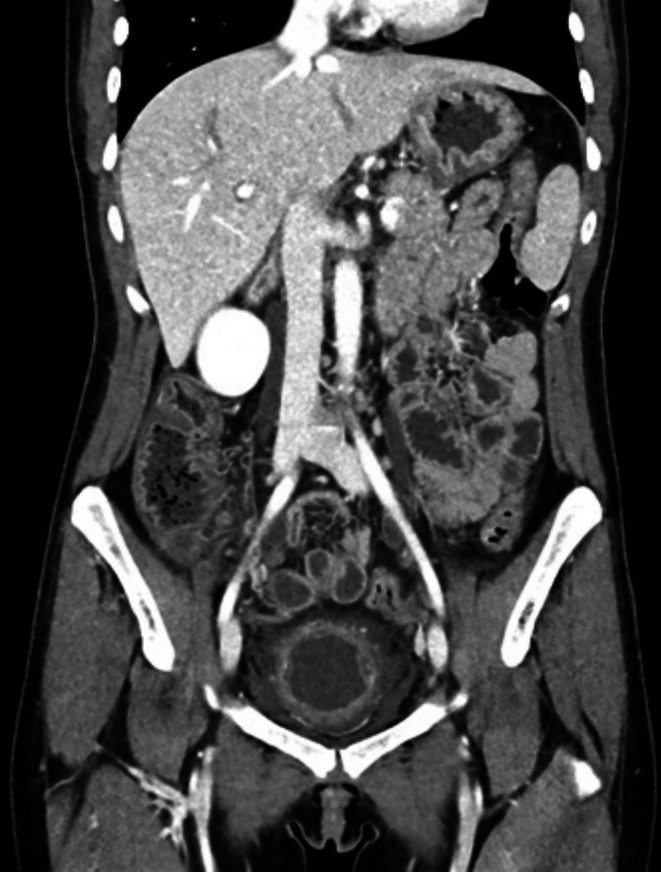 FIGURE 2
FIGURE 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSalivary Gland Disorders and Functions · Liver Diseases and Immunity · Celiac Disease Research and Management
Summary
- Sjögren's syndrome can present with recurrent, unexplained gastrointestinal symptoms mimicking other autoimmune diseases; clinicians should consider Sjögren's syndrome when standard GI workups are inconclusive.
Introduction
1
Sjögren's syndrome (SS) is a chronic multisystem autoimmune illness of the exocrine glands that is distinguished by lymphocytic infiltration of the afflicted gland [1]. It primarily affects females in a 9:1 female‐to‐male ratio, occurring across all ages but most commonly in the fourth and sixth decades [2]. The pathogenesis involves lymphocytic infiltration and destruction of the glandular tissue, thus reducing the glandular secretion. Salivary and lacrimal glands are the most frequently involved, with significant loss of their secretory function resulting in oral and eye dryness [2, 3]. Systemic manifestations, occurring in approximately 30% to 40% of patients, can affect the lungs, kidneys, vasculature, and blood [2, 4]. The gastrointestinal manifestation of SS primarily affects the upper gastrointestinal tract, resulting in dyspepsia, chronic atrophic gastritis, and primary biliary cirrhosis. Our case describes an unusual SS case with small intestine involvement, which is considered to be very rare [5].
Case History and Examination
2
This 27‐year‐old woman was diagnosed with SS based on elevated antinuclear antibodies (ANA), rheumatoid factor (RF), positive anti‐SSA antibodies, and lip biopsy. Lupus‐specific serologies, including anti‐dsDNA and anti‐Smith antibodies, were negative, making systemic lupus erythematosus (SLE) unlikely. She initially received hydroxychloroquine but discontinued it after 1–2 months due to severe headaches. Two years after her diagnosis, she began experiencing cyclic episodes of nausea and vomiting, resulting in an inability to tolerate oral intake and significant weight loss, which prompted her visit to our emergency department. A computed tomography (CT) scan of the abdomen and pelvis raised concerns about pancolitis and terminal ileitis (Figure 1). Due to concern for an inflammatory etiology, the gastroenterology team was consulted, and the patient underwent esophagogastroduodenoscopy (EGD) and colonoscopy. EGD showed grade A esophagitis, erythematous mucosa, gastric mucosal atrophy, and a normal duodenum. Colonoscopy showed small nonbleeding internal hemorrhoids; otherwise, it was unremarkable. Her biopsies were unrevealing. She was subsequently discharged on pantoprazole, bowel regimen, and anti‐nausea medications.
CT scan prior to therapy: bladder edema, pancolitis, and terminal ileitis with mucosal hyperenhancement of the bowel and moderate ascites.
Differential Diagnosis, Investigations, and Treatment
3
A month later, she was readmitted with similar complaints. A repeat CT scan revealed the progression of colitis with increased bowel edema. She developed peritoneal signs and underwent paracentesis, which showed an elevated total nucleated cell count with increased polymorphonuclear cells (PMNs). She was treated with a 5‐day course of intravenous antibiotics. The workup during this admission included a transthoracic echocardiogram, liver biopsy, and gastrointestinal pathogen panel, all of which were unremarkable. The CT angiography of the abdomen showed widely patent celiac, superior mesenteric, and inferior mesenteric arteries, as well as widely patent mesenteric, splenic, and portal veins. Her stool test was positive for Helicobacter pylori . She was discharged with a treatment plan for Helicobacter pylori , including metronidazole, bismuth, doxycycline, and a proton pump inhibitor.
Unfortunately, a month after discharge, her symptoms recurred, leading to another hospitalization. A CT scan during this admission revealed marked intestinal dilation with improving ascites. A differential diagnosis of intestinal angioedema was proposed by the Allergy and Immunology department. Due to concern for angioedema and reduced complement levels, her C1 esterase was checked, which was within normal limits. A full‐thickness biopsy with partial bowel resection was considered, but general surgery was deemed too risky due to significant edema. She underwent a positron emission tomography (PET) scan, which was unrevealing. She was started on an empirical trial of Icatibant as a diagnostic test to explore whether the bradykinin pathway was involved. This made no meaningful difference in her symptoms or CT imaging. After discussion with multiple services, it was decided to initiate a trial of high‐dose steroids, which led to a gradual improvement in symptoms and imaging (Figure 2). She was subsequently discharged on a tapering regimen of prednisone, 40 mg daily.
CT scan after therapy: decreased mural thickening, decreased ascites, and decreased bladder wall edema.
She had just tapered her prednisone dose from 10 mg to 5 mg when she began experiencing a recurrence of her cyclic pain, nausea, and emesis, prompting a visit to the emergency department. A CT scan revealed nonspecific gastritis and colitis with gastric wall and distal colonic edema, showing improvement from previous imaging. The Hepatology team considered porphyria as a possible diagnosis, and a genetic panel revealed abnormalities, with further workup recommended. Laboratory workup revealed decreased complement levels and thrombocytopenia, both of which improved with steroid treatment. Hematology and Oncology noted that a repeat evaluation for porphyria was nondiagnostic and instead attributed her thrombocytopenia to immune thrombocytopenic purpura (ITP). Given that her symptoms appeared to correlate with steroid tapering, it was recommended that she be discharged on prednisone 60 mg daily. Additionally, Rheumatology recommended initiating rituximab to treat her unspecified autoimmune condition.
Conclusion and Results (Outcome and Follow‐Up)
4
After starting rituximab, the patient gradually improved and had no further hospitalizations. Her symptoms became more stable, although she continued to experience some abdominal pain and nausea, especially postprandially, along with mild fatigue. With rituximab, she was able to taper her steroid dosage slowly without major setbacks. As of now, her symptoms have significantly improved, though not completely resolved, and she remains stable on a reduced daily prednisone dose of 5 mg.
Discussion
5
This case may represent the first reported instance of bowel enteritis in SS, a condition more commonly associated with lupus. While enteritis is a well‐documented manifestation in lupus, its occurrence in SS is uncommon, making this a unique and noteworthy finding. Although lupus enteritis was considered in the differential, our patient had negative anti‐dsDNA and anti‐Smith antibodies and lacked systemic features of lupus such as nephritis, cutaneous involvement, or serositis. These findings, together with her established SS diagnosis and steroid/rituximab responsiveness, make lupus enteritis unlikely. This case highlights the need for further investigation into potential gastrointestinal manifestations of SS and broadens our understanding of its systemic involvement. The patient's recurrent hospitalizations and extensive diagnostic workup illustrate the diagnostic complexity of such atypical presentations. While her biopsy results were inconclusive, imaging studies revealed severe inflammation characterized by bladder edema, pancolitis, terminal ileitis, and significant ascites. Her marked improvement with high‐dose steroids and rituximab further illustrates the importance of immunosuppressive therapies in managing gastrointestinal complications of SS, reinforcing the need for aggressive treatment in severe cases.
This case is consistent with previously reported instances of rare gastrointestinal involvement in Crooks and Zweiman, who described a patient with severe nausea, vomiting, and vague abdominal pain, later attributed to renal tubular acidosis, another rare SS manifestation. The patient's symptoms responded favorably to steroid therapy [6]. Similarly, Xu et al. documented a case of a 66‐year‐old man whose primary complaint was chronic refractory diarrhea [7]. Despite normal endoscopic and imaging findings, his diagnosis was confirmed by positive anti‐SSA antibodies, Schirmer's test, and a salivary gland biopsy [7]. Glucocorticoid treatment led to substantial improvement, demonstrating the critical role of immunosuppression in managing gastrointestinal manifestations of SS.
Another multicenter randomized cohort study by Melchor et al. provides critical insights into the prevalence of digestive disorders in patients diagnosed with Sjögren's syndrome (SS) [8]. Among the 437 patients studied, nearly a third (29.5%) experienced atrophic gastritis, 16.9% had esophageal motility dysfunction, 4.2% developed lymphocytic colitis, and 21.1% were diagnosed with autoimmune hepatitis. Furthermore, the study identified a significant correlation between gastrointestinal symptoms and female patients (p = 0.0009), particularly those with comorbid autoimmune disorders such as hypothyroidism and C3 hypocomplementemia (p = 0.040) [8]. These findings show the diverse and systemic nature of SS, highlighting its potential to cause significant gastrointestinal complications.
Sobhani et al. also reported significant remission in a patient presenting with chronic diarrhea, malabsorption, and weight loss following a diagnosis of SS [9]. The patient had diffuse T lymphocytic infiltration throughout the gastrointestinal tract, with symptoms dramatically improving after immunosuppressive therapy. This case, along with others, highlights the importance of early recognition and appropriate treatment of gastrointestinal involvement in SS.
Effective management of these patients relies on prompt diagnosis and the use of immunosuppressive therapies, such as rituximab and high‐dose steroids, to prevent recurrent hospitalizations and manage severe manifestations. This emphasizes the broader importance of considering SS in differential diagnoses when gastrointestinal symptoms are present and the potential benefits of immunosuppressive treatment in achieving long‐term symptom control.
Author Contributions
Oudai Sahwan: conceptualization, writing – original draft, writing – review and editing. Fares Jamal: writing – original draft, writing – review and editing. Amani Elshaer: writing – review and editing. Lin Batha: writing – original draft, writing – review and editing. Tala Shahin: writing – review and editing. Michael M. Pham: supervision, writing – review and editing.
Ethics Statement
The authors have nothing to report.
Consent
Written consent was obtained from the patient for publication.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1R. I. Fox , “Sjogren's syndrome,” Lancet 366, no. 9482 (2005): 321–331.16039337 10.1016/S 0140-6736(05)66990-5 · doi ↗ · pubmed ↗
- 2S. S. Kassan and H. M. Moutsopoulos , “Clinical Manifestations and Early Diagnosis of Sjogren Syndrome,” Archives of Internal Medicine 164, no. 12 (2004): 1275–1284.15226160 10.1001/archinte.164.12.1275 · doi ↗ · pubmed ↗
- 3B. Qin , J. Wang , Z. Yang , et al., “Epidemiology of Primary Sjogren's Syndrome: A Systematic Review and Meta‐Analysis,” Annals of the Rheumatic Diseases 74, no. 11 (2015): 1983–1989.24938285 10.1136/annrheumdis-2014-205375 · doi ↗ · pubmed ↗
- 4X. Mariette and L. A. Criswell , “Primary Sjogren's Syndrome,” New England Journal of Medicine 378, no. 10 (2018): 931–939.29514034 10.1056/NEJ Mcp 1702514 · doi ↗ · pubmed ↗
- 5E. C. Ebert , “Gastrointestinal and Hepatic Manifestations of Sjogren Syndrome,” Journal of Clinical Gastroenterology 46, no. 1 (2012): 25–30.22157220 10.1097/MCG.0b 013e 3182329 d 9c · doi ↗ · pubmed ↗
- 6G. W. Crooks and B. Zweiman , “An Unusual Presentation of Sjogren's Syndrome,” Clinical and Diagnostic Laboratory Immunology 3, no. 4 (1996): 483–484.8807219 10.1128/cdli.3.4.483-484.1996 PMC 170377 · doi ↗ · pubmed ↗
- 7L. Xu , M. Gui , C. Sun , V. Yau , C. Sun , and J. Qi , “Refractory Diarrhea in a Patient With Sjogren's Syndrome: A Case Report,” Frontiers in Nutrition 10 (2023): 1086967.36824167 10.3389/fnut.2023.1086967 PMC 9941144 · doi ↗ · pubmed ↗
- 8S. Melchor , C. Sanchez‐Piedra , M. Fernandez Castro , et al., “Digestive Involvement in Primary Sjogren's Syndrome: Analysis From the Sjogrenser Registry,” Clinical and Experimental Rheumatology 38, no. Suppl 126 (2020): 110–115.33025900 · pubmed ↗