Causal relevance of clonal haematopoiesis with cardiac disease and adverse remodelling: a Mendelian randomisation study
Alec Peter Morley, Maddalena Ardissino, Paul Carter, Betty Raman, Adam J Mead, Pedro M Quiros, George S Vassiliou, Zahra Raisi-Estabragh

TL;DR
This study uses genetic data to suggest that clonal haematopoiesis may cause heart disease and structural heart changes, offering new insights into potential early interventions.
Contribution
The study is the first to use Mendelian randomization to link clonal haematopoiesis with adverse cardiac magnetic resonance phenotypes and atrial fibrillation.
Findings
DNMT3A-CH and small-clone-CH are associated with increased atrial fibrillation risk.
CH is linked to larger heart chamber sizes and signs of myocardial fibrosis.
TET2-CH is associated with higher myocardial native T1 time, indicating possible fibrosis.
Abstract
Many observational studies highlight clonal haematopoiesis (CH) as a novel determinant of cardiovascular disease (CVD). However, disentangling cause and effect from important confounders, such as age and smoking, is challenging. Mendelian randomisation (MR) was used to assess the causal relationships of CH with (1) major CVD outcomes associated with adverse remodelling, and (2) cardiovascular magnetic resonance (CMR) phenotypes which have not been examined previously. Uncorrelated (r2<0.001), genome-wide significant (p<5×10−6) single nucleotide polymorphisms were extracted from Genome-Wide Association Study summary statistics for CH (any subtype), gene-specific CH subtypes (DNMT3A and TET2), and CH clonal size subtypes (small clone and large clone). Mendelian Randomisation using a Robust Adjusted Profile Score (MR-RAPS) was used for analyses on outcomes of atrial fibrillation (AF),…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
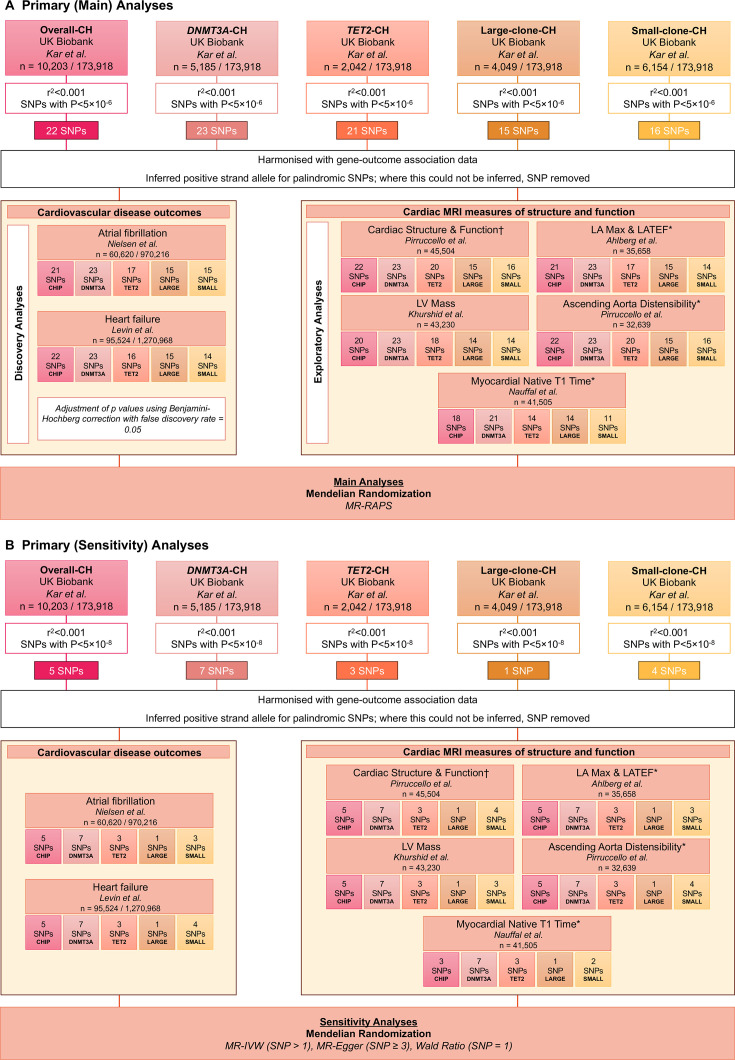 Figure 1
Figure 1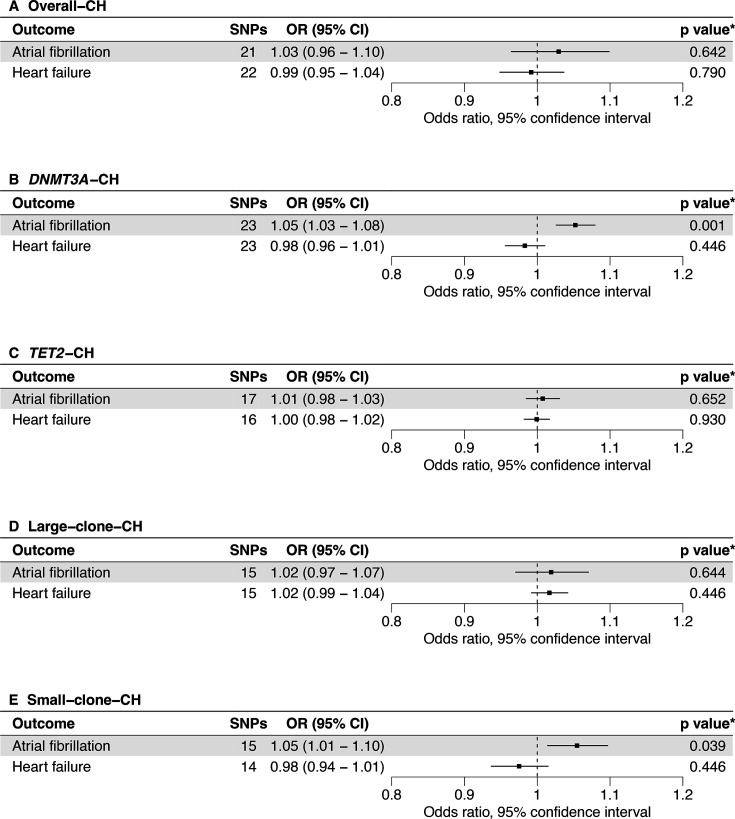 Figure 2
Figure 2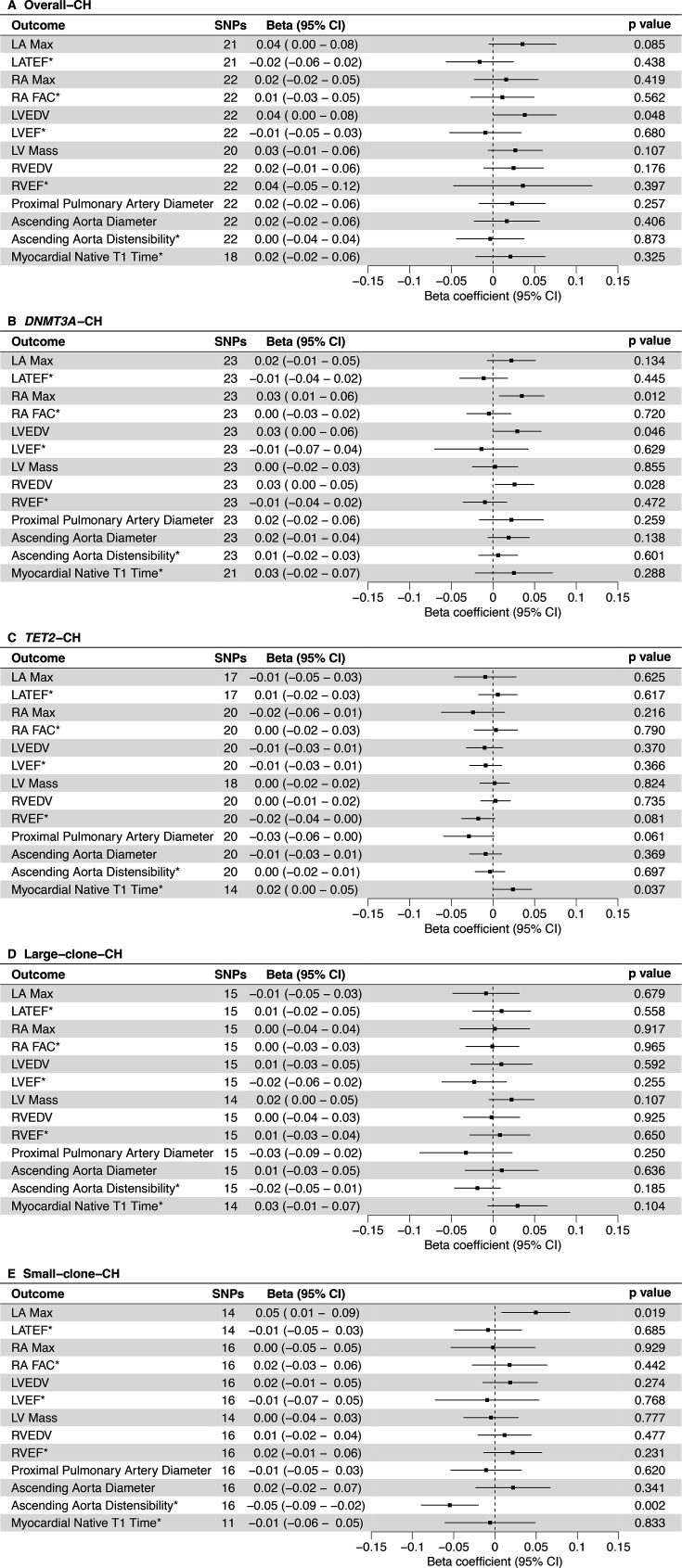 Figure 3
Figure 3| Phenotype | Study or consortium | Ancestry | Cases/controls | Case definition | Control definition | GWAS units | PMID |
|---|---|---|---|---|---|---|---|
| Exposures for primary analysis | |||||||
| Overall-CH | Kar | EUR | 10 203/173 918 | Any CH | No CH | Log(OR) | 35 835 912 |
| | Kar | EUR | 5185/1 73 918 | CH | No CH | Log(OR) | 35 835 912 |
| | Kar | EUR | 2042/173 918 | CH | No CH | Log(OR) | 35 835 912 |
| Large-clone-CH | Kar | EUR | 4049/173 918 | CH | No CH | Log(OR) | 35 835 912 |
| Small-clone-CH | Kar | EUR | 6154/173 918 | CH | No CH | Log(OR) | 35 835 912 |
| Exposures for replication analysis | |||||||
| Overall-CH | Kessler | EUR | 25 657/342 869 | CH | No CH | Log(OR) | 36 450 978 |
| | Kessler | EUR | 16 219/342 869 | CH | No CH | Log(OR) | 36 450 978 |
| | Kessler | EUR | 3918/342 869 | CH | No CH | Log(OR) | 36 450 978 |
| Exposures for validation analysis | |||||||
| Systolic blood pressure | Evangelou | EUR | 757 601 | N/A | N/A | mmHg | 30 224 653 |
| Outcomes | |||||||
| Atrial fibrillation | Nielsen | EUR | 60 620/970 216 | Clinically diagnosed atrial fibrillation or flutter | No history of atrial fibrillation, flutter or other arrhythmias | Log(OR) | 30 061 737 |
| Heart failure | Levin | EUR | 95 524/1270 968 | Diagnosis of heart failure by physician, or healthcare record, and corroborated on self-report | No history of heart failure | Log(OR) | 36 376 295 |
| Cardiac structure and function | Pirruccello | EUR | 45 504 | UK Biobank participants | N/A | 1-SD | 35 697 867 |
| Left atrial maximum volume and left atrial total ejection fraction | Ahlberg | EUR | 35 658 | UK Biobank participants | N/A | 1-SD | 34 338 756 |
| Left ventricular mass | Khurshid | EUR | 43 230 | UK Biobank participants | N/A | 1-SD | 36 944 631 |
| Ascending aorta distensibility | Pirruccello | EUR | 32 639 | UK Biobank participants | N/A | 1-SD | 37 019 578 |
| Myocardial native T1 time | Nauffal | EUR | 41 505 | UK Biobank participants | N/A | 1-SD | 37 081 215 |
- —http://dx.doi.org/10.13039/501100004895European Social Fund Plus
- —http://dx.doi.org/10.13039/100014440Ministerio de Ciencia, Innovación y Universidades
- —Ramon y Cajal Program
- —http://dx.doi.org/10.13039/501100004587Instituto de Salud Carlos III
- —http://dx.doi.org/10.13039/501100000781European Research Council
- —http://dx.doi.org/10.13039/100010269Wellcome Trust
- —http://dx.doi.org/10.13039/501100000272National Institute for Health and Care Research
- —http://dx.doi.org/10.13039/501100000402Kay Kendall Leukaemia Fund
- —http://dx.doi.org/10.13039/501100000289Cancer Research UK
- —http://dx.doi.org/10.13039/501100000265Medical Research Council
- —http://dx.doi.org/10.13039/501100000274British Heart Foundation
- —http://dx.doi.org/10.13039/501100015570Blood Cancer UK
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Myeloid Leukemia Research · RNA modifications and cancer · Myeloproliferative Neoplasms: Diagnosis and Treatment
Background
Ageing is associated with the acquisition of somatic mutations. While most have no functional significance, some somatic mutations provide a cell survival advantage leading to establishment and outgrowth of mutation-bearing clones, with potentially deleterious outcomes. Clonal haematopoiesis (CH) refers to the clonal expansion of individual haematopoietic stem cells and their progeny driven by somatic mutations, in the absence of haematological malignancy.1 Around 70% of CH cases with known leukaemia-associated mutation drivers occur in the genes encoding epigenetic regulators DNMT3A and TET2, with most of the remainder driven by mutations in genes for chromatin regulator ASXL1, DNA damage response proteins PPM1D and P53, splicing factors SF3B1 and SRSF2, or the tyrosine kinase JAK2.2
CH is the precursor of most myeloid neoplasms, but less than 2% of CH carriers develop these cancers with risk varying by mutant gene and clone size.3 This modest elevation in myeloid cancer risk explains only a small part of the increased overall mortality risk associated with CH,4 much of which has been attributed to augmented cardiovascular disease (CVD) susceptibility. Growing evidence from observational studies demonstrates independent associations of CH with a range of CVD outcomes, including atherosclerotic disease,5 atrial fibrillation (AF),6 heart failure (HF)710 and aortic aneurysms.11 The magnitude of these relationships and CH prevalence is comparable to that of traditional risk factors, heralding CH as a novel CVD determinant. Mechanistic studies have implicated inflammatory mechanisms12 related to augmented mutant macrophage inflammation and inflammasome activity. However, others have suggested the associations could be influenced by confounding from major shared risk factors, such as smoking,13 accelerated aging,14 shared genetic risk or reverse causation.15
Mendelian randomisation (MR) is a genetic epidemiological method that leverages the random inheritance of genes to investigate exposure-outcome relationships within the construct of a natural experiment.16 This methodology results in random distribution of confounders independent of genetic risk,17 similar to a randomised study, greatly mitigating against the influence of biases such as reverse causation and unmeasured confounding. Thus, MR offers the opportunity to support causal associations between CH and CVD outcomes, provided certain assumptions are met.
Cardiovascular magnetic resonance (CMR) provides highly detailed organ-level information about health and disease patterns, indicating preclinical disease states and providing insight into potential underlying disease processes.18 19 CMR is the reference modality for evaluation of cardiovascular structure and function, and uniquely provides non-invasive information about myocardial character almost akin to a tissue biopsy.20 CMR has huge potential for risk stratification of people with CH; however, relationships of CH with CMR phenotypes have not been previously assessed with genetic or observational studies, representing an important knowledge gap, which this study aims to address.
This study uses MR to evaluate causal associations of different CH subtypes and clone sizes with: (1) CVD outcomes associated with adverse remodelling (AF and HF), and (2) 13 genetically predicted CMR-derived phenotypes of cardiovascular structure and function. We validate our findings across a range of sensitivity and independent replication analyses.
Methods
This study used publicly available Genome-Wide Association Study (GWAS) summary data available to download at cited sources.2130 A summary of sources is provided in table 1.
For the primary analyses, gene-exposure association estimates were extracted from Kar et al’s21 GWAS with CH variants from the UK Biobank differentiated as (1) overall-CH, (2) DNMT3A-CH, (3) TET2-CH, (4) large-clone-CH and (5) small-clone-CH (figure 1). Gene-outcome association estimates of AF24 and HF25 were used for the discovery analysis, and 13 CMR phenotype estimates2630 were used for the exploratory analysis.
Primary analyses design using exposure instruments from Kar et al.21 CH, clonal haematopoiesis; LA Max, left atrial maximum volume; LATEF, left atrial total ejection fraction; LV Mass, left ventricular mass; MR-Egger, Mendelian randomisation using Egger regression; MR-IVW, Mendelian randomisation using the inverse variance weighted method; MR-RAPS, Mendelian Randomisation using a Robust Adjusted Profile Score; SNP, single nucleotide polymorphism. , not indexed to body surface area; †, ascending aorta diameter, left ventricular end-diastolic volume (LVEDV), left ventricular ejection fraction (LVEF), proximal pulmonary artery diameter, right atrial fractional area change (RA FAC), right atrial maximum area (RA Max), right ventricular ejection fraction (RVEF), right ventricular end-diastolic volume (RVEDV).*
After harmonisation, Mendelian Randomisation using a Robust Adjusted Profile Score31 (MR-RAPS) was used for primary analysis to estimate associations between genetically predicted CH and outcomes, which used relaxed selection criteria of p<5×10^−6^ and r^2^<0.001. Sensitivity analyses were carried out using Mendelian randomisation using the inverse variance weighted method16 (MR-IVW), Mendelian randomisation using Egger regression32 (MR-Egger), and, where appropriate, the Wald ratio method;33 these methods used conventional selection criteria of p<5×10^−8^ and r^2^<0.001.
The primary analysis was split into a discovery phase with CVD outcomes and an exploratory phase with CMR phenotypes. For CVD outcomes, results are presented as an OR with a respective 95% CI. All p values in the discovery analyses were Benjamini–Hochberg34 corrected for multiple testing with a 5% false discovery rate across all exposure-CVD outcome pairs. For CMR phenotypes, results are presented as a beta coefficient (β) with a 95% CI.
Replication analyses were performed using exposure instruments provided by Kessler et al22 from the UK Biobank defined as (1) overall-CH, (2) DNMT3A-CH and (3) TET2-CH (online supplemental figure 1). Furthermore, a validation analysis was conducted using exposure instruments of systolic blood pressure (SBP) from Evangelou et al23 to ensure consistency with recent observational evidence35 and to test the veracity of the approach (online supplemental figure 2). Finally, we conducted a phenome-wide scan of each single nucleotide polymorphism used as an instrumental variable in the analyses, to identify gene-exposure associations with alternative phenotypes.
The full methods for this study can be found in the online supplemental methods.
Results
CH and CVD risk (AF and HF)
The primary discovery analyses using Kar et al’s21 instruments (figure 2) showed that AF risk was increased by DNMT3A-CH (OR 1.05 (1.03 to 1.08), p=8.65×10^−4^) and small-clone-CH (OR 1.05 (1.01 to 1.10), p=3.91×10^−2^). Overall-CH, TET2-CH and large-clone-CH were also directionally associated with increased AF risk but were statistically non-significant. No significant associations were found with HF. Associations were consistent across the sensitivity analyses, with no evidence of directional pleiotropy (online supplemental table 2). Within the sensitivity analyses, we identified additional significant associations of overall-CH with increased AF risk (OR 1.09 (1.04 to 1.15), p=4.90×10^−4^), and DNMT3A-CH with decreased HF risk (OR 0.97 (0.95 to 1.00), p=2.86×10^−2^).
*Primary analysis using MR-RAPS for the effects of CH from Kar et al21 on CVD outcomes. CH, clonal haematopoiesis; CVD, cardiovascular disease; MR-RAPS, Mendelian Randomisation using a Robust Adjusted Profile Score; SNP, single nucleotide polymorphism. , adjusted values of p (false discovery rate = 5%).
The replication analyses using Kessler et al’s22 instruments (online supplemental table 3) directionally supported the primary analyses. No statistically significant associations were identified with any CH subtype and CVD outcome. However, similar to the primary analyses, overall-CH, DNMT3A-CH and TET2-CH were directionally associated with increased AF risk. We identified no significant associations on replication sensitivity analyses and note no evidence of directional pleiotropy.
The validation analyses with increased SBP (by 5 mmHg) confirmed the suitability of our methods (online supplemental table 4) demonstrating significant associations with increased AF and HF risk (online supplemental figure 3).
CH and CMR phenotypes
The exploratory analyses with Kar et al’s21 instruments and CMR phenotypes are shown in figure 3. Overall-CH was associated with increased left ventricular end-diastolic volume (LVEDV) (β 0.04 (0.00 to 0.08), p=4.81×10^−2^). DNMT3A-CH was associated with increased right atrial maximum area (RA Max) (β 0.03 (0.01 to 0.06), p=1.16×10^−2^), LVEDV (β 0.03 (0.00 to 0.06), p=4.57×10^−2^) and right ventricular end-diastolic volume (RVEDV) (β 0.03 (0.00 to 0.05), p=2.79×10^−2^). TET2-CH was associated with increased myocardial native T1 time (β 0.02 (0.00 to 0.05), p=3.71×10^−2^). Small-clone-CH was associated with increased left atrial maximum volume (LA Max) (β 0.05 (0.01 to 0.09), p=1.89×10^−2^) and decreased ascending aorta distensibility (β −0.05 (−0.09 to −0.02), p=1.88×10^−3^). No significant associations with large-clone-CH were identified. Associations were consistent on sensitivity analyses, with no evidence of directional pleiotropy (online supplemental table 5). Within the sensitivity analyses, we identified additional significant associations; overall-CH was associated with increased RVEDV (β 0.06 (0.02 to 0.11), p=5.48×10^−3^) and proximal pulmonary artery diameter (β 0.07 (0.02 to 0.12), p=8.72×10^−3^). TET2-CH was associated with increased LVEDV (β 0.03 (0.00 to 0.07), p=4.58×10^−2^) and proximal pulmonary artery diameter (β 0.05 (0.02 to 0.09), p=3.78×10^−3^). Finally, small-clone-CH was associated with increased LVEDV (β 0.06 (0.01 to 0.10), p=1.36×10^−2^).
*Primary analyses using MR-RAPS for effects of CH from Kar et al21 on CMR phenotypes. Beta, beta coefficient; CH, clonal haematopoiesis; CMR, cardiovascular magnetic resonance; LA Max, left atrial maximum volume; LATEF, left atrial total ejection fraction; LV Mass, left ventricular mass; LVEDV, left ventricular end-diastolic volume; LVEF, left ventricular ejection fraction; MR-RAPS, Mendelian Randomisation using a Robust Adjusted Profile Score; RA FAC, right atrial fractional area change; RA Max, right atrial maximum area; RVEDV, right ventricular end-diastolic volume; RVEF, right ventricular ejection fraction; SNP, single nucleotide polymorphism. not indexed to body surface area.
The replication analyses, using Kessler et al’s22 instruments, were directionally supportive of the primary exploratory analyses. These were also consistent on sensitivity analyses, and we note no evidence of directional pleiotropy (online supplemental table 6). Furthermore, the sensitivity analyses identified an association of DNMT3A-CH with increased RA Max (β 0.05 (0.01 to 0.09), p=1.80×10^−2^), RVEDV (β 0.04 (0.01 to 0.07), p=1.60×10^−2^) and ascending aorta diameter (β 0.03 (0.00 to 0.05), p=3.14×10^−2^). Finally, TET2-CH was associated with increased LVEDV (β 0.04 (0.01 to 0.07), p=8.04×10^−3^), LV Mass (β 0.04 (0.01 to 0.07), p=2.92×10^−3^) and RVEDV (β 0.04 (0.01 to 0.07), p=6.77×10^−3^).
The validation analyses with increased SBP (by 5 mmHg) confirmed method suitability (online supplemental table 7) demonstrating significant associations with increased LA Max, LVEDV, LVEF, LV Mass, RVEF and ascending aorta diameter, and associations with decreased LATEF, ascending aorta distensibility and myocardial native T1 time (online supplemental figure 4).
Phenome-wide analyses
The results of the phenome-wide analyses are presented in online supplemental table 8.
Discussion
CH has been widely associated with heightened CVD risk in observational analyses,511 supported by experimental models of DNMT3A36 37 and TET253740 mediated CH. Although extremely useful for mechanistic insight, such models often reflect rare and more extreme CH phenotypes, with effects on CVD outcomes diminished, or even lost, with heterozygotic mutations, making extrapolation difficult to human populations with a greater heterozygotic burden.41 Also, the close relationship between ageing and both CH and CVD makes it difficult to eliminate residual confounding in observational studies, particularly as chronological age adjustment cannot fully account for impacts of biological age.14 Therefore, causal links between CH and CVD remain uncertain.
Using Mendelian randomisation methods to mitigate residual confounding and reverse causation, our study triangulates previous observational results and provides new evidence supporting potential causal associations of CH with AF. We further extend existing evidence, using CMR-derived phenotypes to characterise, for the first time, adverse cardiovascular remodelling patterns associated with CH; our findings indicate genetic associations with larger atrial and ventricular sizes, higher LV mass, higher myocardial native T1 time, and lower aortic distensibility, although in a subtype specific manner. The robustness of our methods and findings is demonstrated across multiple validation and replication analyses.
Amongst subtypes examined, DNMT3A-CH exhibited the largest and most consistent associations with AF risk and adverse cardiovascular remodelling. This may indicate heterogenic associations with CVD across CH subtypes, with dominance of individual driver genes conferring differing downstream consequences. This is supported by weaker, less consistent associations for overall-CH, a composite category of mutants, despite greater statistical power. However, this is unsurprising given driver genes have differing functional roles, with sometimes biologically opposing functions. For example, the key function of DNMT3A is DNA methylation leading to transcription repression,42 whereas TET2 is an important epigenetic regulator, causing DNA demethylation and widespread gene activation.43 Despite this, studies have found a paradoxical convergence of DNMT3A and TET2-mediated effects.5 36 Although we did not find significant associations between TET2-CH and AF, this contradicts previous observational studies;39 44 Ahn et al44 demonstrated heightened AF risk in individuals with TET2-CH and large-clone-CH. Given that TET2-CH is associated with increased AF risk across sensitivity analyses and that there are fewer individuals within this subtype, lack of associations within the primary analyses is very likely to be due to limited power.
Atrial remodelling is integral in arrhythmia predisposition and AF pathogenesis—which frames the key finding of DNMT3A-CH and small-clone-CH being concomitantly associated with AF and larger atrial size. Given higher proportions of DNMT3A mutants in small-clone-CH versus large-clone-CH subtypes within the primary analysis21 instruments (53% DNMT3A, 20% TET2 mutants in small clones; 46% DNMT3A; 21% TET2 mutants in large clones), these similar findings are expected and support previous findings of increased atrial size44 and AF incidence for CH subtypes.6 Increased atrial size robustly associates with heightened AF risk45 46 and is an established precursor to AF onset,47 with dilatation and fibrosis associated with conduction irregularity. However, AF itself can lead to atrial dilatation48 which further begets AF in a vicious spiral. Mechanistically, CH may predispose individuals to atrial dilatation and myopathy which may occur alongside other risk factors. In an angiotensin-II hypertension murine model,37 DNMT3A and TET2 mutations induced structural and functional changes similar to those described. Inflammatory cytokines are increasingly recognised as integral in driving cardiac remodelling and AF risk. Increased high-sensitivity C reactive protein (hs-CRP) has been identified in CH,49 and associations with AF are attenuated on hs-CRP50 adjustment, suggesting a potential mediating role. Furthermore, Nlrp3 inflammasome activation has been proposed to promote arrhythmogenesis through altered cardiomyocyte calcium handling in mice with TET2 mutations.38 Notably, in the current study, TET2-CH was associated with an increase in myocardial native T1 time, a proxy of myocardial fibrosis associated with CVD risk,51 and has been described in observational studies of UK Biobank participants.52 Although not yet studied in DNMT3A-CH experimental models, Nlrp3 inflammasome activating mutations have been found to have convergent effects to TET2 mutations in macrophages,36 suggesting a similar phenomenon may occur. We highlight this as an area for further investigation.
HF has been implicated as a consequence of CH in observational studies and experimental models71053 which is suggested to be driven by similar inflammatory mechanisms to those previously discussed. Generally, our analyses did not find significant associations between any CH subtype and HF, which is supported by some previous observational studies710 and MR analyses.21 In one sensitivity analysis, we found a lower rate of HF with DNMT3A-CH, which may be explained by survival bias among the outcome GWAS25 given that previous observational analyses have demonstrated increased mortality in DNMT3A-CH-driven HF.7 DNMT3A-CH did, however, associate with increased LVEDV and RVEDV, with some suggestion of similar volumetric changes and increased myocardial T1 time for *TET2-*CH. These structural changes may represent early compensatory changes in preclinical HF,54 55 and the benefits of lower imaging threshold in these individuals warrant investigation.
The strength of our study arises from leveraging large-scale GWAS summary statistics for exposures and outcomes. Furthermore, we use MR-RAPS,31 which can increase statistical power by including conventionally ‘weaker instruments’ and overcome current challenges in CH investigations. Similar CH investigations56 have used this approach and, with consideration of MR-RAPS limitations, can provide evidence supporting causality.31 Using such genetic inferential techniques helps to support and validate current literature which has described observational associations between CH subtypes and adverse remodelling.52 Finally, the validation analyses support our approach with associations consistent with current knowledge of SBP as a risk factor for AF57 58 and HF,59 alongside recent observational evidence which corroborates findings of adverse CMR phenotypes.35
Limitations
While helpful in removing systematic biases, MR cannot completely exclude the potential for horizontal and/or vertical pleiotropy,60 although appreciation of relevant potential biological mechanisms can aid in understanding these limitations. An obvious limitation is that telomere-mediated biological ageing can be argued to influence CH and outcome associations. Telomere lengthening61 has been associated with CH subtypes, whereas shortening is associated with CVD.62 However, to cause significant pleiotropy, the CH and CVD relationships with telomere length would need to be directionally consistent. Moreover, the lack of significant MR-Egger intercepts suggests significant pleiotropy is unlikely,32 although our findings should be interpreted with this in mind. The accuracy of our results further rests on the definition within the original publications21 22 and recognise this remains controversial.63
We note that significant associations were not replicated in the study by Kessler et al,22 which could result from regression dilution. Additionally, we acknowledge that results from the study by Kar et al21 may be interpreted as ‘random positive’, although we argue this is unlikely given biological and directional consistency of results. Associations with TET2 were limited, and we acknowledge that current literature with observational studies64 has identified TET2-CH to be associated strongly with HF. Insufficient power is the most likely explanation, given that subtypes with significant associations had higher instrument strength. A repeated analysis leveraging targeted CH sequencing is likely to yield more insights, as our ability to detect the impact of smaller clones is limited with whole-exome sequencing;63 we are unable to do this at present given the lack of publicly available summary statistics of such data, and highlight this as an area for further investigation. In addition, genetics-based inferential studies are vulnerable to instrumental variable assumption violations, which we mitigate by careful instrument selection and by using a variety of MR methods, but we note this cannot be fully eliminated. For example, to address the first instrumental variable assumption (that variants must be able to predict the exposure), we calculate F-statistics to identify weak instruments. We did not find any variants with an F-statistic <10, providing no evidence of weak instruments. Furthermore, in order to reduce violations of the second instrumental variable assumption (no common causes of the genetic variant and the outcome), this analysis is limited to individuals of European ancestry to reduce confounding from population stratification. However, this does mean that validation in other groups is needed, given that we are unable to extrapolate our findings to populations of other ancestries. Finally, although we use a single data set for exposures and outcomes (the UK Biobank), recent evidence suggests risk of biased estimates due to correlation is minimal when using two-sample MR methods in large data sets.65
Conclusions
This study supports potential causal associations between DNMT3A-CH and small-clone-CH with AF risk and atrial dilatation. Furthermore, we identify, for the first time, associations of DNMT3A-CH with left and right ventricular dilatation and of *TET2-*CH with increased myocardial native T1 time which may represent manifestations of preclinical HF. These structural changes may indicate a potential window of opportunity for intervention, such as by risk stratification and early preventative strategies to improve patient outcomes. Our findings extend the growing literature regarding the cardiovascular sequelae of CH and structural abnormalities. Further research is required to establish the pathophysiological mechanisms underlying increased CVD risk and their clinical implications, and the best interventions to attenuate such risk.
Supplementary material
10.1136/openhrt-2025-003602online supplemental file 1
10.1136/openhrt-2025-003602online supplemental table 1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Steensma DP Bejar R Jaiswal S et al Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes Blood 201512691610.1182/blood-2015-03-63174725931582 PMC 4624443 · doi ↗ · pubmed ↗
- 2Marnell CS Bick A Natarajan P Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease J Mol Cell Cardiol 20211619810510.1016/j.yjmcc.2021.07.00434298011 PMC 8629838 · doi ↗ · pubmed ↗
- 3Gu M Kovilakam SC Dunn WG et al Multiparameter prediction of myeloid neoplasia risk Nat Genet 20235515233010.1038/s 41588-023-01472-137620601 PMC 10484784 · doi ↗ · pubmed ↗
- 4Jaiswal S Fontanillas P Flannick J et al Age-related clonal hematopoiesis associated with adverse outcomes N Engl J Med 201437124889810.1056/NEJ Moa 140861725426837 PMC 4306669 · doi ↗ · pubmed ↗
- 5Jaiswal S Natarajan P Silver AJ et al Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease N Engl J Med 20173771112110.1056/NEJ Moa 170171928636844 PMC 6717509 · doi ↗ · pubmed ↗
- 6Saadatagah S Naderian M Uddin M et al Atrial Fibrillation and Clonal Hematopoiesis in TET 2 and ASXL 1JAMA Cardiol 2024949750610.1001/jamacardio.2024.045938598228 PMC 11007653 · doi ↗ · pubmed ↗
- 7Assmus B Cremer S Kirschbaum K et al Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT 3A- and TET 2-driver gene mutations Eur Heart J 2021422576510.1093/eurheartj/ehaa 84533241418 · doi ↗ · pubmed ↗
- 8Yu B Roberts MB Raffield LM et al Supplemental Association of Clonal Hematopoiesis With Incident Heart Failure J Am Coll Cardiol 202178425210.1016/j.jacc.2021.04.08534210413 PMC 8313294 · doi ↗ · pubmed ↗