A mechanism underlying the association of Aβ plaque and lipid droplets in Alzheimer's disease
Lixuan Ren, Xiwen Ma, Jianping Ye

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
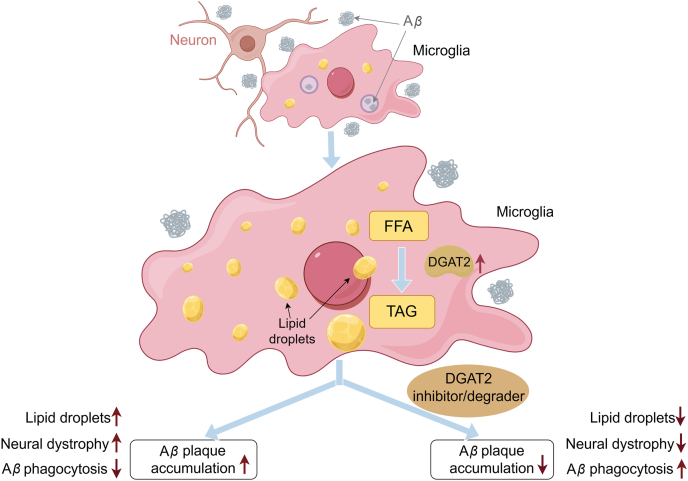 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAlzheimer's disease research and treatments · Metabolomics and Mass Spectrometry Studies · Neurological Disorders and Treatments
The mechanism of lipid droplet (LD) formation in glial cells is an active area in the field of Alzheimer's disease (AD) pathology. A recent study titled “Amyloid-β induces lipid droplet-mediated microglial dysfunction via the enzyme DGAT2 in Alzheimer's disease” by Prakash et al.1 is published in Immunity to report a role of Aβ protein in the induction of LD formation in microglia for the phagocytosis dysfunction in AD. In this context, diacylglycerol O-acyltransferase 2 (DGAT2) is identified as a potential therapeutic target for its role in LD formation.
AD is characterized by amyloid-β (Aβ) plaque accumulation as a result of microglia failure in clearance of the plaques. Although microglial dysfunction in phagocytosis is known to contribute to Aβ plaque formation, the underlying mechanisms remain unclear for the phagocytosis defect. LD accumulation in microglia is a pathological character of AD, which was first reported by Dr. Alzheimer together with the Aβ plaques in the brains of AD patients. However, the significance of LD accumulation was largely overlooked in the AD field for nearly a century, until recent findings identified the apolipoprotein APOE4 as a genetic risk factor for AD. Microglia with LD accumulation and transition into a dysfunctional state are known as lipid droplet-accumulating microglia (LDAM). LDAM are increased in AD brains and enriched in individuals with the APOE4/4 genotype. Emerging research has uncovered a connection between genetic risk factors and accumulation of LDs in microglia, which results in production of neurotoxic factors from the microglia2^,^3. APOE4 increases LD formation by interrupting lipid metabolism, and thereby impairing neuronal function in the mechanism of cognitive decline4^,^5. The new study demonstrates that Aβ promotes LD formation by induction of lipid-synthesizing enzyme DGAT2, which in turn impairs the phagocytic activity of microglia.
The study first revealed that LD accumulation was increased in microglia of 5×FAD mice, in which Aβ accumulation is increased at 2–3 months of age by the transgene, and cognitive function is impaired after 6 months in age6. LD accumulation occurred after Aβ accumulation at 5–7 months of age. The degree of accumulation was significantly influenced by age, sex, and brain region. The observation was made by comparing the neutral lipid content in microglia isolated from the mouse brain using flow cytometry. The AD mice exhibited a substantial increase in cerebral LDs at 5–7 months of age, with female mice showing a greater increase. The difference was not observed in younger mice at 3–4 months of age. The data suggest that LD accumulation occurs after Aβ deposition in the 5×FAD mice.
The distribution of LD was investigated in multiple brain regions using label-free stimulated Raman scattering (SRS) microscopy. Hippocampal sections showed a higher LD burden in 5×FAD female mice compared to wild-type (WT) controls, with numerous LDs distribution around Aβ plaques. This observation was confirmed by immunohistochemical co-staining (methoxy X04 for amyloid plaques, LipidTox for lipids, IBA1 for microglia). Similar patterns were observed in the cortex and hippocampal subfields (CA1 and subiculum) of 5×FAD females. Across these regions, LD ^+^ microglia were located mostly around Aβ plaques. In the subiculum, microglia adjacent to plaques demonstrated an amoeboid morphology with shorter processes. Correspondingly, LD^+^ microglia were mostly clustered nearby the plaques, with density declining away from the plaque cores. A parallel analysis of postmortem hippocampal tissue of AD patients demonstrated a significantly higher LD density compared to the non-AD individuals. The LD-laden microglia were substantially elevated in the hippocampi of AD patients, consistent with the findings in the mouse model.
Microglial phagocytosis of Aβ is a critical mechanism for plaque clearance7. To investigate whether direct exposure to Aβ would impair the phagocytic function in microglia, microglia were isolated from the mice and treated in cell culture with Aβ^pH^—a pH-dependent fluorescent probe that emits green fluorescence in acidic lysosomes upon phagocytosis. Subsequently, the cells were analyzed using flow cytometry. The results showed that although both 5×FAD and WT microglia exhibited an increase in LD content in response to the Aβ exposure, only the 5×FAD microglia exhibited a reduction in phagocytic activity (by 40%).
To investigate whether Aβ directly induces the LD formation, the WT microglia were treated with Aβ in cell culture for varying durations at 1, 12, and 24 h. All intracellular and extracellular lipids were quantified and characterized by mass spectrometry-based multiple reaction monitoring (MRM) profiling. After 1 h of treatment, the free fatty acids (FFAs) showed the most significant changes, particularly very-long-chain saturated FFAs such as C20:0, C22:0, and C19:0. However, when the treatment duration reached 24 h, triglycerides (TGs) exhibited the most notable changes instead of FFAs. Overall, FFAs initially increased at the beginning, while TG levels rose at 24 h. Pathway analysis revealed that Aβ activated the glycerol phosphate and monoacylglycerol pathways, promoting TG synthesis. This metabolic reprogramming was associated with a reduction in phagocytic capacity in the microglia. These findings indicate that Aβ protein directly promotes the formation of LDs in microglia by inducing lipogenesis.
At the molecular level, the genes underlying the Aβ-induced lipogenesis were investigated. DGAT2 was identified as a key enzyme activated by Aβ exposure in the induction of LD accumulation. DGAT2 is a metabolic enzyme that catalyzes the conversion of diacylglycerol (DAG) into TGs in the cytosol. Elevated levels of DGAT2 protein were observed in LD-laden microglia in both 5×FAD mouse brains and AD patient brains. Inhibition of DGAT2 (D2i) reduced LD formation and restored phagocytic function of microglia in Aβ clearance. To suppress the DGAT2 activity in vivo, a protein degrader targeting DGAT2 was infused into the brain at lateral ventricles of 11- to 24-month-old 5×FAD mice. Animals that received the drug for a period of 1 week showed a significant reduction in the Aβ plaque burden, which was observed by comparing the treated and untreated groups. The plaque reduction was approximately 51% in the subiculum region of the hippocampus and LD reduction was 40% in LD-positive microglia near plaques.
This study proposed a molecular model, in which Aβ exposure leads to the accumulation of FFAs in microglia, and DGAT2 converts the FFAs into TGs, resulting in LD formation. This metabolic rewiring impairs phagocytic activity of microglia, promoting the Aβ plaque accumulation. The study also explored the therapeutic potential of targeting DGAT2. Inhibition of DGAT2, either through gene knockdown or protein degradation, showed promise in restoring microglial function and reducing plaque burden—even at advanced stages of AD. However, since DGAT2 is expressed in various cell types, systemic inhibition could lead to serious side effects. Cell type-specific degradation of DGAT2 in the brain, as demonstrated in preclinical models, may effectively avoid the systemic complications.
The study has several limitations. First, the impact of the findings on cognitive function was not assessed in the mouse model, which limits the overall significance of the results. Second, other potential molecules involved in LD formation were not investigated. Third, the mechanism of phagocytosis inhibition by LD was not investigated. Additionally, the mechanisms underlying the FFA induction by Aβ exposure, as well as the effects on other cell types, remain unexplored.
In conclusion, this study demonstrates that Aβ induces LD accumulation in microglia by promoting triglyceride synthesis through DGAT2 activation. The resulting LD buildup impairs microglial phagocytic function, thereby exacerbating Aβ plaque formation in AD. Inhibiting DGAT2 activity may represent a promising therapeutic strategy to restore microglial function in Aβ clearance, ultimately reducing plaque deposition in the brain (Fig. 1).Figure 1. Pathway of Aβ induction of lipid droplets in microglia.Figure 1
Author contributions
Lixuan Ren drafted the manuscript. Xiwen Ma and Jianping Ye provided the idea and revised the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Prakash P.Manchanda P.Paouri E.Bisht K.Sharma K.Rajpoot J.Amyloid-β induces lipid droplet-mediated microglial dysfunction via the enzyme DGAT 2 in Alzheimer's disease Immunity 582025153615524039345410.1016/j.immuni.2025.04.029PMC 12168635 · doi ↗ · pubmed ↗
- 2Haney M.S.Pálovics R.Munson C.N.Long C.Johansson P.K.Yip O.APOE 4/4 is linked to damaging lipid droplets in Alzheimer's disease microglia Nature 62820241541613848089210.1038/s 41586-024-07185-7PMC 10990924 · doi ↗ · pubmed ↗
- 3Mao M.Ma X.Wang X.Ye J.Microglial APOE 4 promotes neuron degeneration in Alzheimer's disease through inhibition of lipid droplet autophagy Acta Pharm Sin B 1520256576604004192410.1016/j.apsb.2024.10.009PMC 11873644 · doi ↗ · pubmed ↗
- 4Jeong W.Lee H.Cho S.Seo J.Apo E 4-induced cholesterol dysregulation and its brain cell type-specific implications in the pathogenesis of Alzheimer's disease Mol Cell 42201973974610.14348/molcells.2019.0200 PMC 688397931711277 · doi ↗ · pubmed ↗
- 5Sienski G.Narayan P.Bonner J.M.Kory N.Boland S.Arczewska A.A.APOE 4 disrupts intracellular lipid homeostasis in human i PSC-derived glia Sci Transl Med 132021 eaaz 456410.1126/scitranslmed.aaz 4564 PMC 821859333658354 · doi ↗ · pubmed ↗
- 6Oakley H.Cole S.L.Logan S.Maus E.Shao P.Craft J.Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation J Neurosci 26200610129101401702116910.1523/JNEUROSCI.1202-06.2006 PMC 6674618 · doi ↗ · pubmed ↗
- 7Leng F.Edison P.Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here?.Nat Rev Neurol 1720211571723331867610.1038/s 41582-020-00435-y · doi ↗ · pubmed ↗