The locus coeruleus maintains core body temperature and protects against hypothermia during dexmedetomidine-induced sedation
Berta Anuncibay Soto, Ying Ma, Mathieu Nollet, Sara Wong, Giulia Miracca, Daniel Rastinejad, Raquel Yustos, Alexei L. Vyssotski, Nicholas P. Franks, William Wisden

TL;DR
This study shows that the locus coeruleus helps protect against hypothermia during DEX sedation in mice, challenging previous assumptions about its role.
Contribution
The study reveals that the locus coeruleus maintains body temperature during DEX sedation and is not essential for inducing NREM-like sleep.
Findings
The locus coeruleus maintains body temperature via the preoptic hypothalamus during DEX sedation.
Mice without functional locus coeruleus neurons experience deeper hypothermia when given DEX.
The locus coeruleus is not required for DEX-induced NREM-like sleep but aids in recovery from hypothermia.
Abstract
Dexmedetomidine (DEX), a widely used sedative in intensive care, induces an arousable state resembling non-rapid eye movement (NREM) sleep and lowers body temperature. For some patients, even slight decreases in body temperature during sedation could pose health risks. It is commonly believed that DEX targets inhibitory adrenergic receptors on norepinephrinergic locus coeruleus (LC) neurons to induce sedation. However, our study in mice challenges this. We found that the LC, via the preoptic hypothalamus, helps maintain body temperature. Without this LC–hypothalamus link, mice are more sensitive to DEX, resulting in deeper hypothermia. The LC is also not needed for the induction of NREM-like sleep by DEX. On the other hand, the LC accelerates recovery from hypothermia and may reduce adverse outcomes related to sedation. Activating the adra2a receptors of the brain’s norepinephrine (NE)…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
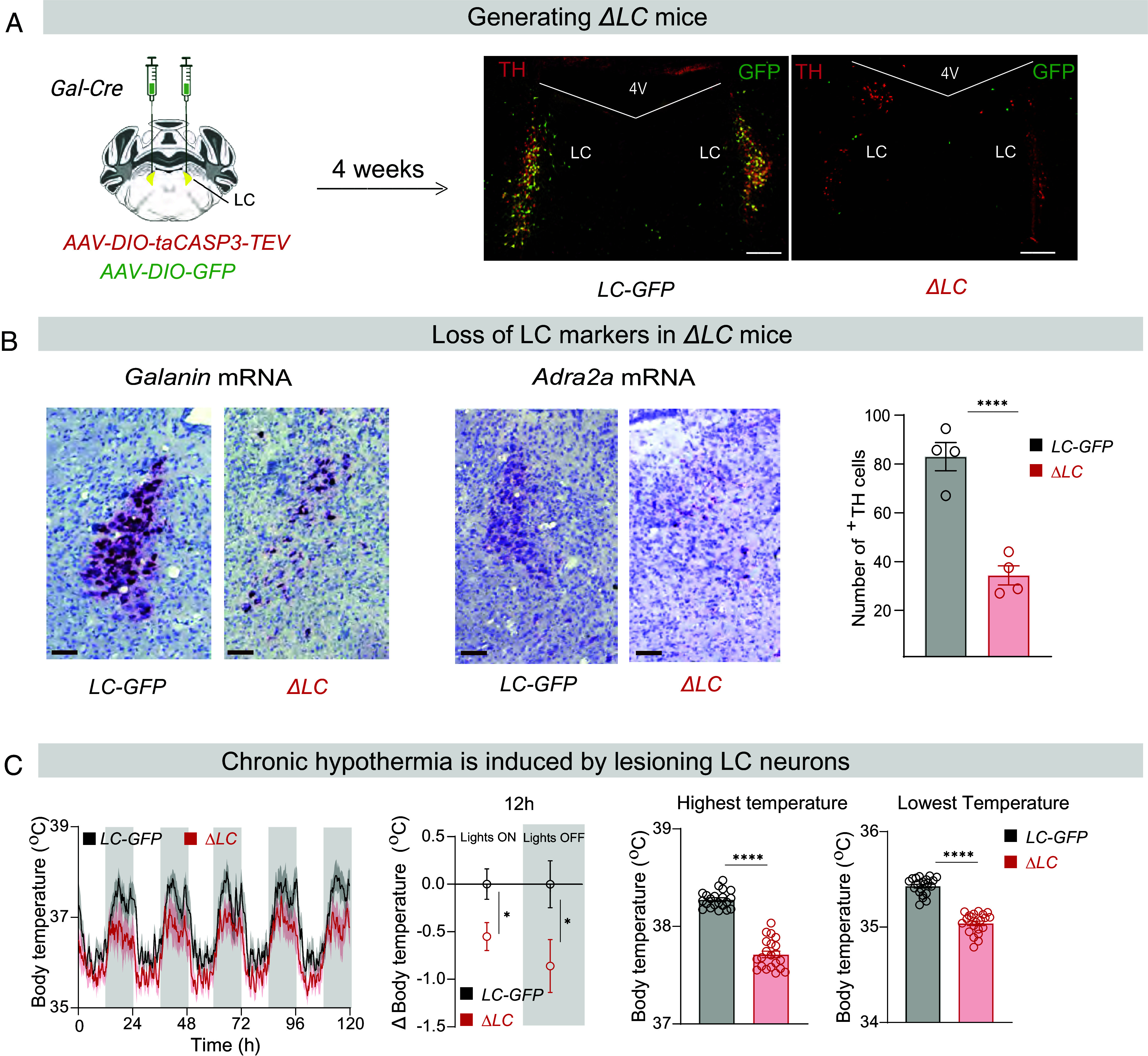 Fig. 1
Fig. 1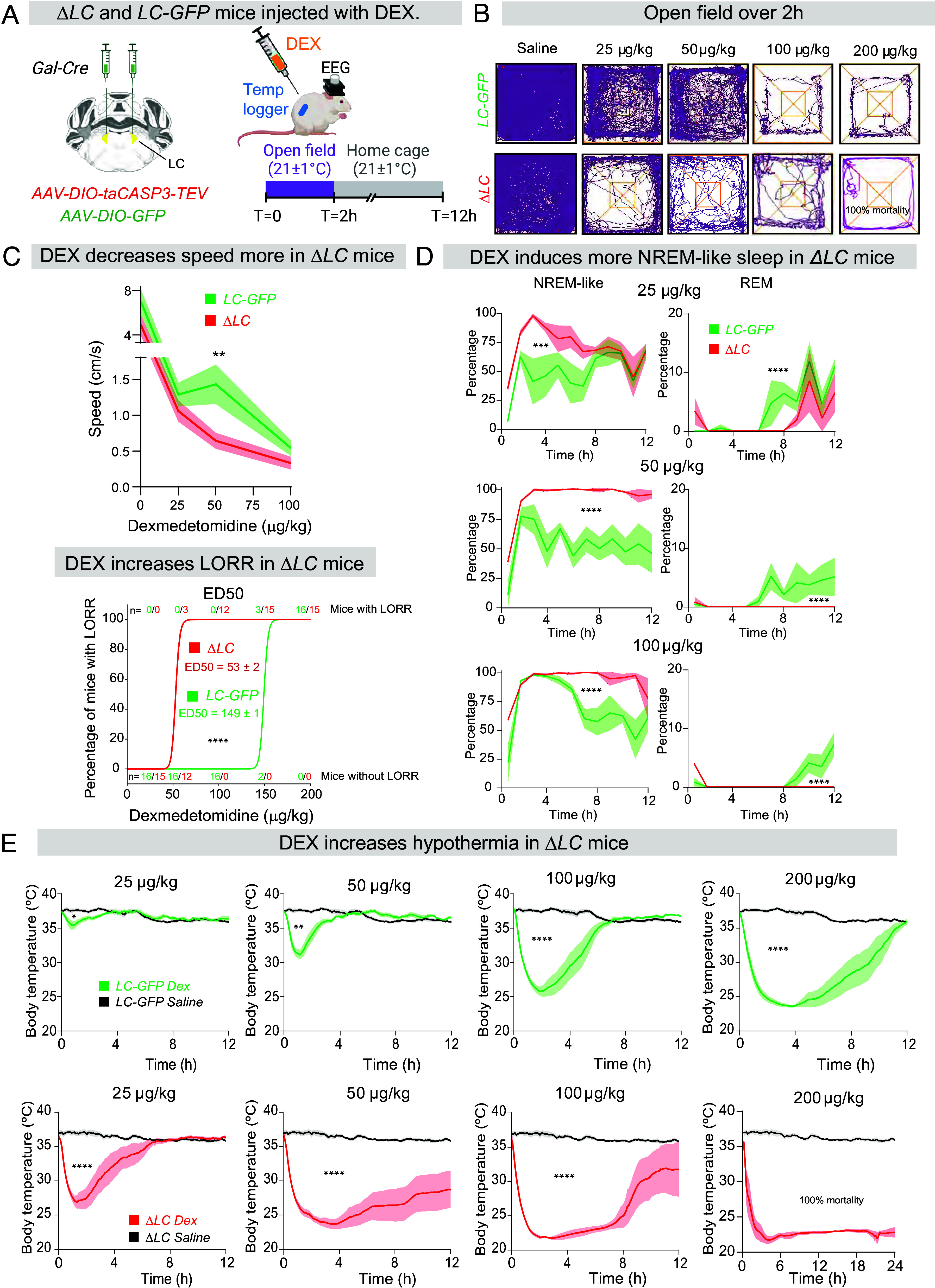 Fig. 2
Fig. 2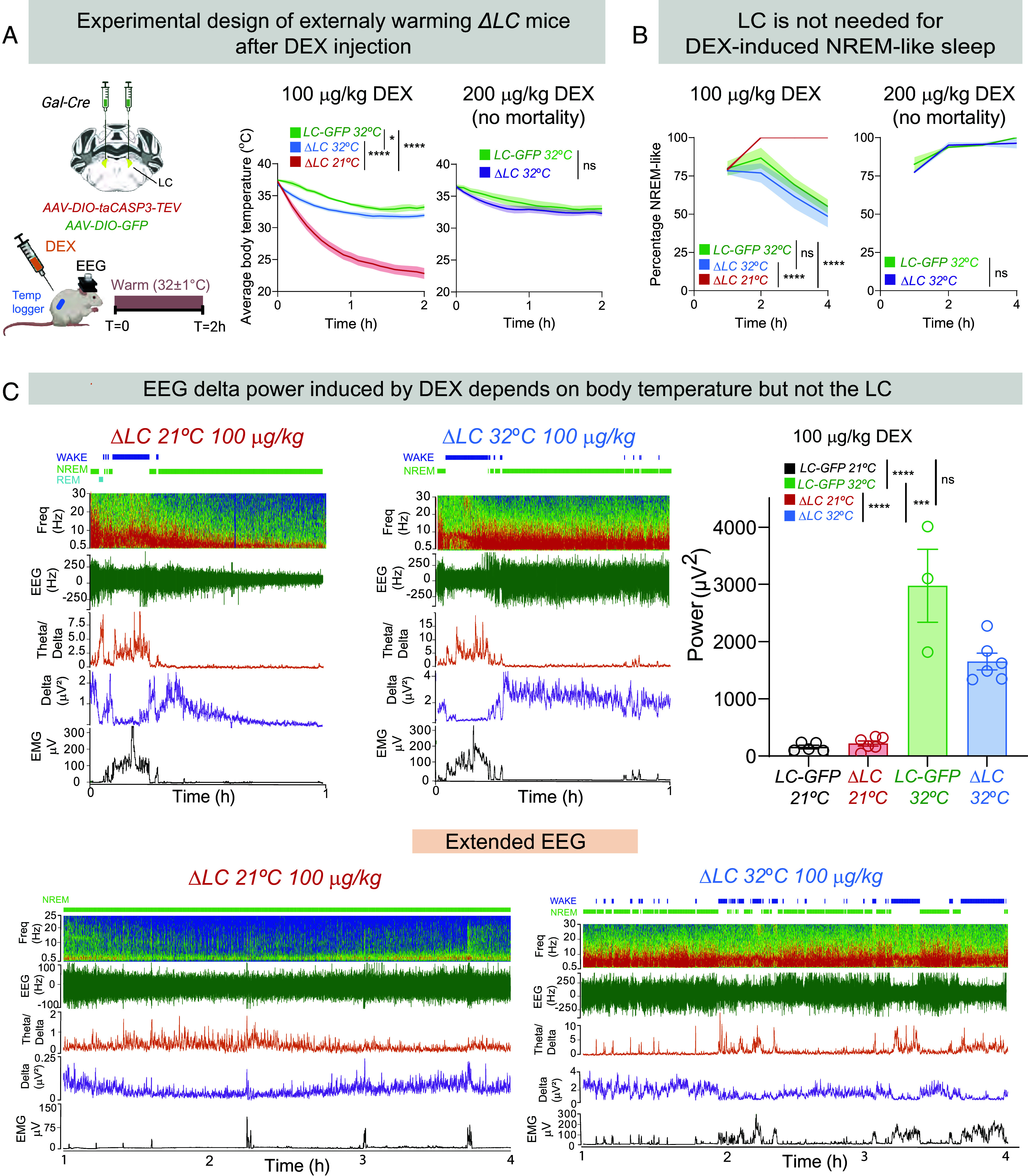 Fig. 3
Fig. 3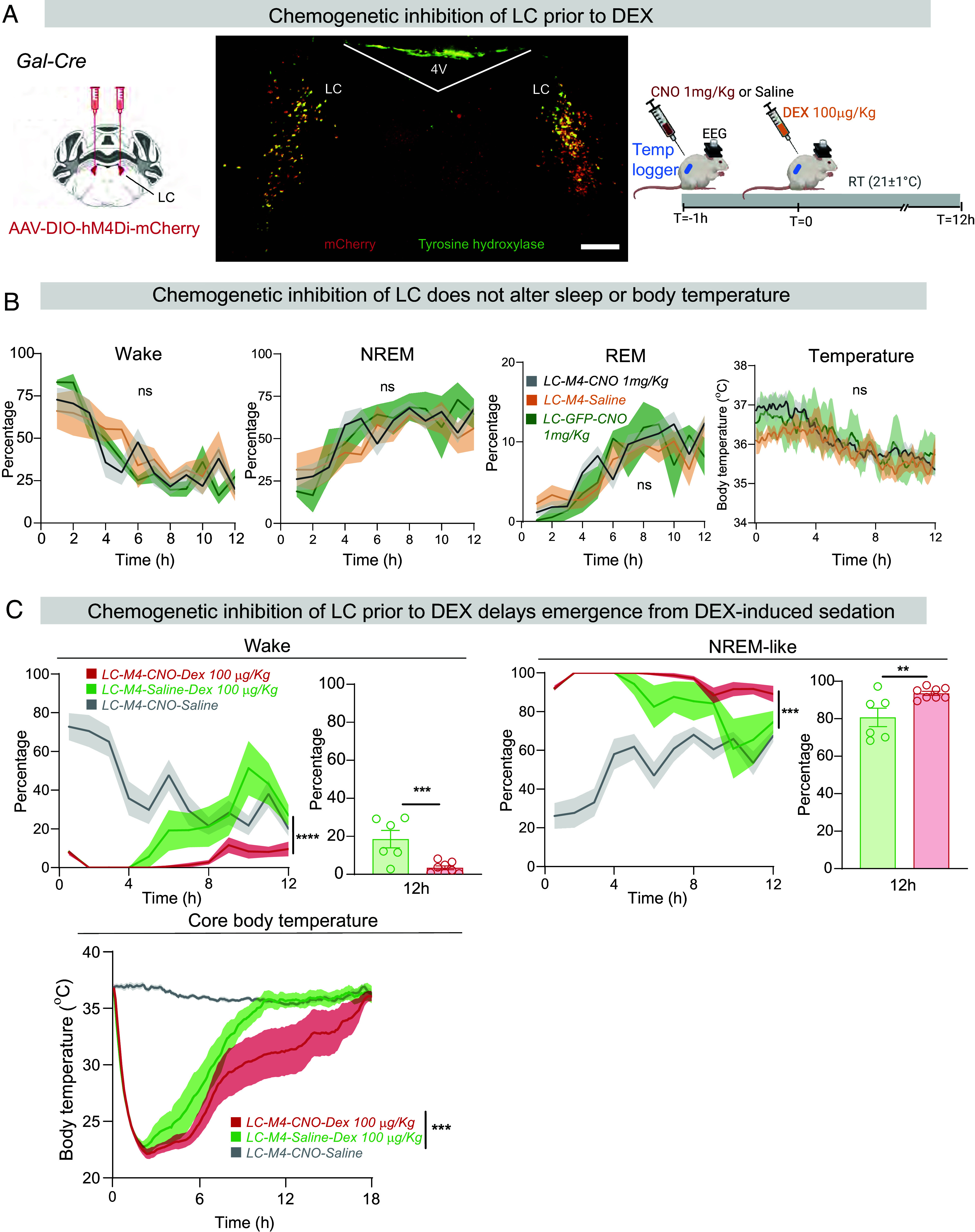 Fig. 4
Fig. 4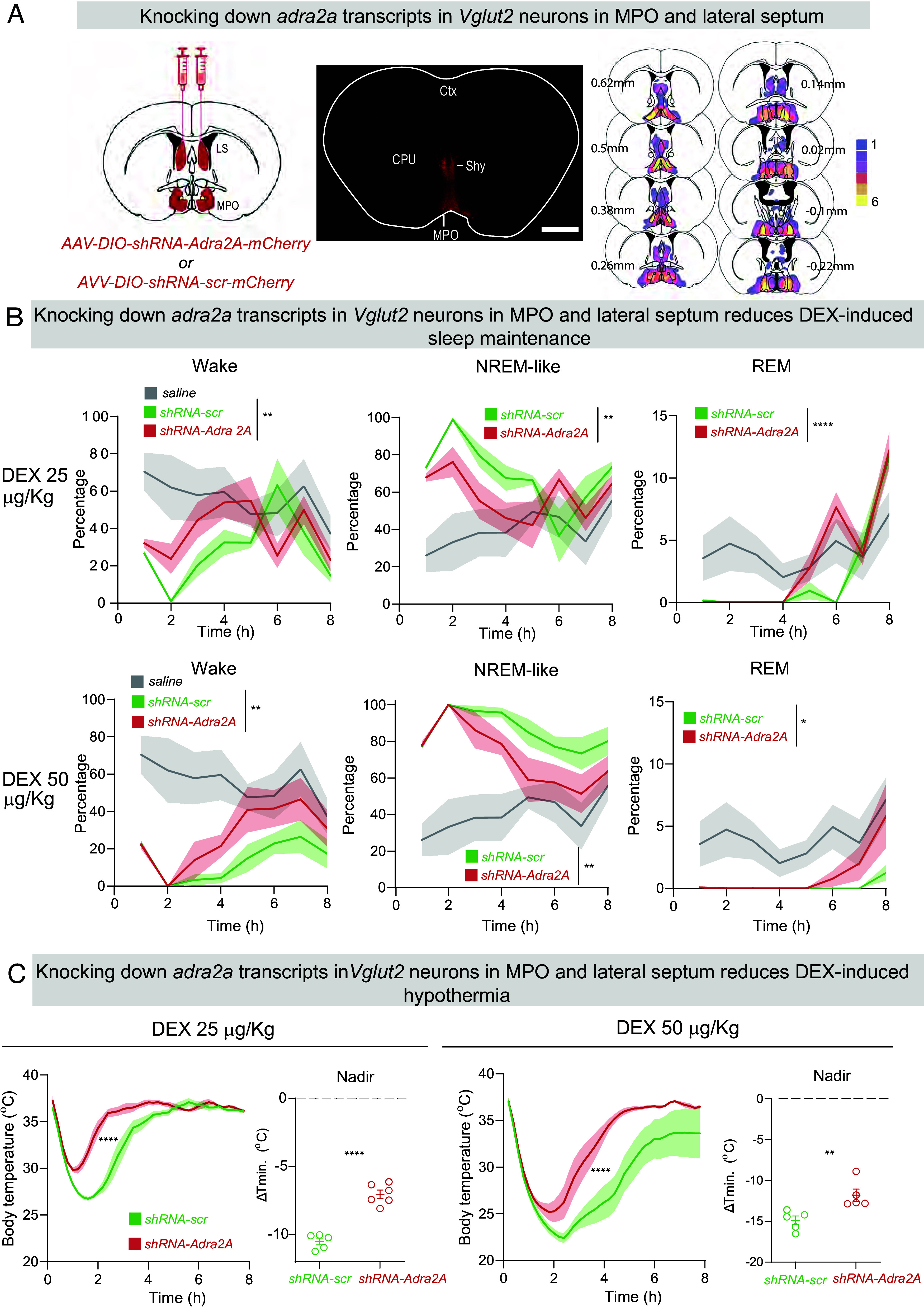 Fig. 5
Fig. 5- —Wellcome Trust (WT)100010269
- —UK Dementia Research Institute (UK DRI)501100017510
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsThermal Regulation in Medicine · Anesthesia and Neurotoxicity Research · Intensive Care Unit Cognitive Disorders
Norepinephrine (NE) is usually associated with arousal, but one branch of the NE system, when selectively activated, produces NREM-like sleep and hypothermia (1). This is exemplified by dexmedetomidine (DEX), an α2 receptor adreno-agonist (2). DEX, an important intensive care drug, produces arousable sedation with a reduced risk of delirium (3????–8), and is also widely used in veterinary medicine (9). Electroencephalogram (EEG) measurements show the sleep state induced by DEX in humans resembles stage 2 and 3 NREM sleep, with delta (1 to 4 Hz) waves (10??–13). In rodents, DEX similarly produces NREM-like sleep (14??????–21); and REM sleep is suppressed by DEX in both humans and rodents (10, 18, 19). Further highlighting the similarity between DEX-induced and natural NREM sleep, changes in regional cerebral blood flow evoked by DEX in humans mirror those in natural NREM sleep (22).
In animals and humans, in addition to inducing a NREM-like sleep state, DEX lowers body temperature, and heart and metabolic rates (14?–16, 21, 23?–25). Indeed, without external warming the state induced by DEX can be viewed as torpor-like (21). During anesthesia and sedation, although every attempt is made to keep patients warm, inadvertent hypothermia (defined clinically as a core body temperature less than 35 °C) is a complication for some patients (26, 27). While hypothermia is beneficial for cardiac surgery and organ transplantation (27), consequences of inadvertent hypothermia can include increased blood loss, increased risk of wound infections and poorer healing, myocardial ischemia, and postoperative cognitive dysfunction (26, 28, 29). The latter may be caused by increased phosphorylation of the microtubule-associated protein tau, which in turn could increase the risk of developing neurodegenerative disease (28, 30). Early after DEX was introduced clinically, it was suggested that the drug disrupts body temperature control, causing an expanded temperature range that does not trigger thermoregulatory responses (31). Thus, it was proposed that patients sedated with DEX might have an increased risk of hypothermia (31). Although hypothermia is not usually a clinical complication of DEX, there have been cases where DEX caused mild hypothermia in patients (32, 33).
Total gene knockouts in mice show that adra2a receptors mediate DEX’s sedative and hypothermic actions (23, 34). Where in the brain are these adra2a receptors that DEX works on? From analyzing the relative timing of delta oscillations in the neocortex and thalamus, DEX initiates NREM-like sleep subcortically (20). Much of the brain’s NE is produced in widely projecting neurons in the locus coeruleus (LC)/group A6 in the brainstem (35). The dominant theory for how DEX induces a NREM sleep-like state is that it binds to adra2a autoreceptors on locus ceruleus (LC) neurons to reduce NE release throughout the brain (3, 36??–39). For example, direct infusion of a high concentration (165 mM) of DEX into the rat LC induced behavioral sedation as defined by loss-of-righting reflex (38), and indeed, we have found in acute slice recordings that DEX inhibits spontaneous activity of LC cells via adra2a receptors (18).
Direct agonist infusion into the LC does not rule out the drug reaching other brain regions via diffusion through the nearby 4th ventricle, however. In rats with NE transmission removed by toxin depletion of NE stores, DEX could still lower the minimum alveolar concentration of halothane needed for anesthesia, suggesting DEX acts postsynaptically of the LC to induce sedation (40). Indeed, we have found by c-fos activity tagging, that neurons in the mouse PO hypothalamic area are sufficient for DEX-induced NREM sleep and hypothermia (18). Furthermore, within the lateral PO (LPO) hypothalamic area, genetically lesioning galanin peptide (gal)-expressing neurons reduced DEX’s ability to induce NREM-like sleep and hypothermia (19). But although LPO gal neurons and other hypothalamic neurons near the supraoptic nucleus (SON), as well as neurons in the suprachiasmatic nucleus, are part of the circuitry by which DEX acts to induce NREM-like sleep (18, 19, 41, 42), these cells themselves might not express the critical adra2a receptors. Instead, the prime movers for initiating DEX’s sleep and hypothermic actions could still be adra2a receptors expressed on the LC neurons. To test this hypothesis, we selectively lesioned the LC and examined how mice responded to DEX.
Strikingly, without the LC, mice were more hypothermic at baseline and more sensitive to DEX, showing prolonged NREM-like sleep and deeper hypothermia. These effects depended on LC projections to the medial (M) preoptic (PO) hypothalamus, where adra2a receptors are expressed. When ΔLC mice were kept warm, DEX induced similar NREM-like sleep to controls, but with higher delta power. We conclude that the LC is not required for DEX-induced NREM-like sleep and hypothermia. Genetic knockdown showed that adra2a receptors on MPO hypothalamus glutamate cells contribute to DEX’s ability to induce hypothermia. We suggest that NE released from the LC chronically regulates adra2a receptor levels in the PO hypothalamus, the consequence being that an intact LC restrains DEX-induced hypothermia and aids recovery from hypothermia.
Results
Genetic Lesioning of LC Cells to Generate ΔLC Mice.
Galanin (gal) gene expression marks about 80% of norepinephrinergic [tyrosine hydroxylase (TH)-positive] cells in the LC (43????–48). Thus, to ablate the LC, we specifically lesioned Gal-expressing neurons in the LC of adult Gal-Cre mice (Fig. 1A). For the experimental group, AAV-DIO-Caspase and AAV-DIO-GFP viruses were mixed and injected bilaterally into the LC of Gal-Cre mice to give ΔLC mice (Fig. 1A). For controls, AAV-DIO-GFP was injected bilaterally into the LC of Gal-Cre mice to generate LC-GFP mice (Fig. 1A). Four weeks after injection, caspase expression had killed approx. 70% of GFP/TH-expressing cells (see bar graph, Fig. 1 A and B and SI Appendix, Fig. S1A), substantially reducing gal and adra2a mRNA content in the LC (Fig. 1B). The anatomical characterization of the lesion is shown in SI Appendix, Fig. S1A.
*Genetic lesioning of LC neurons produces hypothermia. (A) Top, Schematic of genetic ablation of LC galanin neurons to produce ΔLC mice (Left) and demonstration of ablation of TH cells (Right). LC, locus ceruleus; 4 V, 4th ventricle. (Scale bar, 1,000 µm.) (B) RNAscope images showing loss of galanin and adra2a mRNAs in the LC region of ΔLC mice; bar graph, quantification of ablation of TH cells. Unpaired two-tailed t test, ****P < 0.001 (n = 4 in each group). [Scale bar, 100 µm (brown).] (C) Lesioning of LC neurons reduces body temperature as assayed over multiple days (n = 7, ΔLC and n = 7, LC-GFP control mice); middle, average decrease in body temperatures of ΔLC mice compared with LC-GFP controls (n = 7, unpaired t test, *P < 0.05); and Right, bar graphs of the highest and lowest temperatures varying over the 24 h cycle, (n = 7 per group, 20 highest or lowest temperature reached over 5 d) Unpaired two-tailed t test, ***P < 0.001.
Baseline Sleep–Wake States Are Unchanged, but Body Temperature Is Decreased in ΔLC Mice.
ΔLC mice had the same amounts of wake, NREM and REM sleep as control mice, and there was no change in the frequency of transitions between the three vigilance states (SI Appendix, Fig. S1B). However, there were significant decreases in the maxima and minima of the diurnal core body temperature profiles of ΔLC mice recorded over 5 d compared with the control LC-GFP mice (Fig. 1C). During the 12 h periods of “lights-off” (mice more active) and “lights-on” (mice less active), ΔLC mice were about 1 °C cooler (average core body temperature) in “lights-off” and approx. 0.5 °C cooler during “lights-on” (Fig. 1C); ΔLC mice had clear decreases in both the highest and lowest body temperatures over the diurnal temperature cycle compared with LC-GFP control mice (Fig. 1C).
DEX Potentiates Behavioral Changes in ΔLC Mice.
We examined how DEX induces behavioral changes in ΔLC mice and the LC-GFP controls (Fig. 2A). We tested a range of DEX concentrations, injected i.p., from 25 µg/kg (amount drug/body weight) through to 200 µg/kg. An immediate behavioral difference between ΔLC and LC-GFP controls was apparent. In the open-field test, following DEX injection, ΔLC mice tracked over 2 h were visibly affected by low doses of DEX (25 µg/kg) compared with LC-GFP mice (Fig. 2B), moving more slowly (Fig. 2 C, Upper). (Note: After receiving 200 µg/kg DEX, all the ΔLC mice died, whereas LC-GFP mice survived). The increased sensitivity to DEX of ΔLC mice was also reflected in an approx. threefold left-shift in the quantal dose–response curve for the loss-of-righting reflex (LORR) induced by DEX (Fig. 2 C, Lower panel; DEX ED_50_ for LORR in LC-ΔGal mice, 53 ± 2 µg/kg; and 149 ± 1 µg/kg for LC-GFP mice, measured 90 min after DEX injection).
*Mice with genetically lesioned LC cells are more sensitive to the sleep and hypothermia-inducing properties of DEX. (A) Schematic of genetic ablation of LC neurons to produce ΔLC mice, and experimental design. DEX, dexmedetomidine; EEG, electroencephalogram; LC, locus ceruleus. (B) Example activity traces recorded over 2 h of individual ΔLC and LC-GFP mice in the open-field arena with saline and ascending doses of DEX. (C) Upper graph, speed of ΔLC (n = 7) and LC-GFP (n = 7) mice in the open-field with ascending doses of DEX. Shading indicates SEM (unpaired one tailed t test, **P < 0.01). Lower graph, quantal dose–response curve for LORR with DEX for 15 ΔLC and 16 LC-GFP mice. (****P < 0.001, unpaired two-tailed t test). The numbers above and below each data point indicate the number of mice with (Top row) and without (Bottom row) LORR, respectively, in each group. (D) Amounts of wake, NREM-like sleep, and REM sleep over time following saline or ascending doses of DEX injections in ΔLC (n = 5) and LC-GFP (n = 7) mice (two-way ANOVA, ***P < 0.005; ****P < 0.001). Shading indicates SEM. (E) Kinetics of hypothermia induced by ascending doses of DEX in LC-GFP (Top row, n = 7) and ΔLC (Bottom row, n = 7) mice compared with unchanged body temperatures after saline injection (Shading indicates SEM; two-way ANOVA, *P < 0.05; **P < 0.01; ***P < 0.005; ***P < 0.001).
DEX Induces More NREM-Like Sleep in ΔLC Mice.
We next looked at how DEX affects sleep states (Fig. 2D). Compared with saline i.p. injections of ΔLC and LC-GFP mice, which had no effect on sleep–wake states (SI Appendix, Fig. S1C), DEX i.p. injections produced strong NREM-like sleep, starting 15 min after injection (Fig. 2 D, Left-hand graphs); however, for inducing and sustaining sleep, DEX was more potent in ΔLC mice than in LC-GFP mice (Fig. 2D); in the first 2 to 3 h postinjection, 25 µg/kg DEX already produced 100% NREM-like sleep in ΔLC mice, but for this dose in LC-GFP control mice, sleep was partial (see SI Appendix, Materials and Methods, “Sleep Scoring,” for how we classified the mice as being in a “NREM-like” state, particularly when body temperatures were low). As the DEX dose was increased, 50 µg/kg DEX gave 100% NREM-like sleep for at least 8 h in ΔLC mice, whereas in control mice, again, sleep was more partial. Control mice only achieved 100% NREM-like sleep with 100 µg/kg DEX, but only for the first 4 h (Fig. 2 D, Bottom Left-hand graph). This difference in potency for how DEX induces sedation between the ΔLC mice and LC-GFP mice is also apparent from the perspective that DEX suppresses REM sleep (10). As the DEX is metabolized away, the appearance of REM sleep marks the return of some natural sleep episodes. At 25 µg/kg DEX, LC-GFP control mice experienced their first REM episodes some 6 h postinjection, whereas for ΔLC mice the first REM episodes only started at 8 h postinjection (Fig. 2 D, Right-hand graphs). At higher DEX doses, ΔLC mice never had any REM sleep within 12 h post–DEX injection, whereas for control mice they always experienced REM sleep some 8 h postinjection (Fig. 2 D, Bottom-Right graph). This further illustrates the increased potency of DEX in ΔLC mice to induce and maintain a dominant NREM-like sleep.
DEX Potentiates Hypothermia in ΔLC Mice.
DEX produces hypothermia if animals are not warmed postinjection (18, 19). We looked at how DEX influences body temperature in unwarmed animals at room temperature (21 °C external temp) with, and without, an LC. At 25 µg/kg DEX, control LC-GFP mice had a small 2 °C ± 1.5 °C decrease in core body temperature lasting for 2 h post–DEX injection compared with those mice injected with saline (Fig. 2 E, Top row); however, this dose of DEX in ΔLC mice produced a 10 °C ± 2 °C drop in body temperature lasting around 6 h (Fig. 2 E, Bottom row). As the DEX dose was increased, the extent of hypothermia became more marked in ΔLC mice compared with controls, and it took substantially longer for the core body temperature of ΔLC animals to increase to baseline levels (Fig. 2E). These temperature decreases and recoveries were quantified by measuring the areas (“temperature x time”) formed between the temperature-time curves for saline-injected (baseline) and DEX-injected animals (SI Appendix, Fig. S1D). For ΔLC mice, the amount and persistence of hypothermia induced for any given dose of DEX was much greater than that induced in LC-GFP control mice (SI Appendix, Fig. S1D). At 200 µg/kg DEX, ΔLC mice failed to recover from a sustained hypothermia which persisted for 24 h (Fig. 2E), and there was 100% mortality for this group at this drug dose.
External Warming Allows ΔLC Mice to Respond to DEX in a Similar Way to Controls, and the LC Remains Dispensable for DEX-Induced NREM-Like Sleep.
We tested the effect of DEX in ΔLC mice where the external temperature was maintained at 32 °C for the first 2 h following DEX injection (rather than 21 °C used in the previous experiments) (Fig. 3A). When DEX injections were done with external warming of ΔLC mice at 32 °C, the previously lethal dose of 200 µg/kg DEX was no longer lethal—all ΔLC mice survived. Furthermore, with this initial external warming, DEX at 100 µg/kg now only induced a slight (but still significantly) enhanced hypothermia compared with that induced in LC-GFP control mice (Fig. 3A, compare the green LC-GFP and blue ΔLC curves; the marked hypothermia induced by the same dose of DEX when mice are at ambient temp 21 °C is shown in red). When ΔLC mice with external warming were given 200 µg/kg DEX, their induced hypothermia became further like control LC-GFP mice injected with the same DEX dose [Fig. 3A, were kept for 2 h in a warm environment postinjection, DEX now induced in ΔLC mice a similar amount and time course of NREM-like sleep as seen for control LC-GFP mice (Fig. 3), emphasizing that the LC was not needed for DEX’s sedative actions]. On the other hand, with external warming, less NREM-like sleep was induced in ΔLC mice experiencing 32 °C for the first 2 h following injection than ΔLC mice which were sedated at ambient temperature (Fig. 3 B, Left-hand trace; and Fig. 2D), suggesting that the increased NREM-like sleep in animals maintained only at ambient temperature was a consequence of hypothermia.
*External warming reduces the sensitivity of ΔLC mice to DEX, but the LC remains dispensable for DEX-induced sedation. (A) Schematic of genetic ablation of LC galanin neurons to produce ΔLC mice, and experimental design. EEG, electroencephalogram; LC. locus ceruleus. Right-hand graphs: How external warming (32 °C for 2 h after DEX injection) or room temperature (RT 21 °C) influences the time course of DEX-induced hypothermia in ΔLC (n = 8 at RT; n = 7 at 32 °C) and LC-GFP mice (n = 3 for 32 °C). (two-way ANOVA, *P < 0.05; ****P < 0.001). With external warming after injection, 200 µg/kg DEX is no longer lethal in ΔLC mice. (B) How external warming (32 °C for 2 h after DEX injection) or room temperature (RT 21 °C) influences the time course of DEX-induced NREM-like sleep in ΔLC (n = 8 at RT; n = 6 at 32 °C) and LC-GFP mice (n = 3 for 32 °C). (two-way ANOVA, *P < 0.05; ****P < 0.001). (C) How body temperature affects the DEX-induced EEG delta power independently of the LC. ΔLC (n = 6 at RT; n = 6 at 32 °C) and LC-GFP mice (n = 5 for RT and n = 3 for 32 °C). Example traces showing how environmental warming influences the DEX-induced EEG, and in particular increases the EEG delta power in ΔLC mice for a given dose of DEX (100 µg/kg) over 1 h post–DEX injection. Note the difference in the delta power scales between the 21 °C and 32 °C examples. Bar graph, quantification of DEX-induced (raw) delta power over the 2-h period post–DEX injection at 21 °C and 32 °C in the ΔLC and LC-GFP mice (one-way ANOVA, ***P < 0.005; ***P < 0.001). The lower panel, “Extended EEG,” shows the same examples as above, but extended over 4 h.
DEX-Induced EEG Delta Power Increases with Body Temperature, but the LC Is Dispensable for DEX-Induced Delta Power.
In ΔLC and LC-GFP mice injected at ambient temperatures (21 °C) with DEX, the drug induced a NREM-like EEG classification, but after 2 h the EEG had a low power, likely due to the lower temperatures influencing circuit activity (Fig. 3C) (see SI Appendix, Materials and Methods, “Sleep Scoring,” for how we classified the mice as being in a “NREM-like” sleep during hypothermia”). Over 4 h post–DEX injection, this low delta power was maintained, but the vigilance state still scored as NREM-like sleep (Fig. 3C, “Extended EEG” panel). When the same dose of DEX was given to ΔLC mice in a warm environment, however, an approximately ten times larger delta power was produced that also scored as “NREM-like” sleep (Fig. 3 C, Middle panel and Right-hand bar graphs); this high delta power persisted for several hours (Fig. 3C, “Extended EEG” panel). Thus, these results emphasize that inhibition of the LC by DEX is not required to induce a robust NREM-like sleep state.
LC Activity Enhances Emergence from DEX-Induced Sedation and Hypothermia.
We tested whether transient and reversible inhibition of the LC prior to DEX administration influenced how DEX sedated the mice (Fig. 4A). The inhibitory DREAD receptor hM4di was selectively expressed in the LC by bilaterally injecting AAV-DIO-hM4Di-mCherry into the LC of Gal-Cre mice, generating LC-M4 mice (Fig. 4A). Compared with LC-M4 mice given saline injections or LC-GFP mice given CNO (1 mg/kg i.p.), chemogenetic inhibition (1 mg/kg CNO i.p.) of the LC had no effect on either sleep or core body temperature (Fig. 4B).
*LC activity enhances the emergence from DEX-induced sedation. (A) Schematic of CNO-based chemogenetic inhibition of LC cells prior to DEX administration, and immunocytochemical staining for mCherry and TH showing expression of the hM4Di-mCherry receptors in TH-positive LC cells in a coronal section. 4 V, 4th ventricle. (Scale bar, 1,000 µm.) (B) Chemogenetic inhibition of LC cells with 1 mg/kg CNO but without DEX does not alter baseline sleep–wake or body temperature. Saline + CNO (n = 7) and saline (n = 5) (two-way ANOVA, *P < 0.05). CNO given to LC-GFP animals (n = 5), i.e., no chemogenetic receptor present, served as a further control. (C) Chemogenetic inhibition of the LC delays the emergence of mice from DEX-induced NREM-like sleep and recovery from hypothermia. CNO+saline (n = 12), CNO+Dex (n = 8), and saline+Dex (n = 6) Curves for wake and sleep, two-way ANOVA, ***P < 0.005, ****P < 0.001, Graphs for wake and sleep, unpaired t test one tailed, **P < 0.01, ***P < 0.005; curves for temperature, two-way ANOVA, **P < 0.005.
To assess the effect of preinhibiting the LC prior to DEX administration, CNO or saline was given 1 h before giving 100 µg/kg DEX (Fig. 4A). Induction of NREM-like sleep (at 21 °C external temp) was identical in both groups of mice, as expected if the LC is not needed for DEX to induce NREM-like sleep (Fig. 4C); however, mice with chemogenetically inhibited LC cells, plus subsequent DEX administration (Fig. 4A), slept 4 h longer than mice given DEX alone (Fig. 4C). Following chemogenetic inhibition of the LC, the time course of induction of DEX-induced hypothermia was the same as control mice (Fig. 4C), but the recovery of body temperature was slower. Thus, although the LC is not needed for the induction of NREM-like sleep and hypothermia by DEX, it is required for faster emergence of mice from DEX-induced sleep and hypothermia (Fig. 4C).
The LC Maintains Body Temperature by Projections to the Midline and Medial PO Hypothalamus, and These Intact Projections Reduce the Sensitivity of Mice to DEX.
Given that the PO hypothalamus is a key region controlling body temperature (49) and that the LC sends projections to the hypothalamus (50, 51), we hypothesized that the gal-expressing LC neurons influence body temperature by their projections to the PO. To test this, we injected retro-AAV-DIO-caspase, and retro-AAV-DIO-GFP (as controls), into the MPO and MnPO area of Gal-Cre mice (SI Appendix, Fig. S2A). The retro-AAV capsid ensures that the AAV is taken up by LC terminals and transported back to the cell body (52). In the control mice, approx. 0.85% LC neurons expressed GFP, confirming that a subpopulation of gal-expressing LC cells send projections to the MPO and MnPO areas (SI Appendix, Fig. S2A). In the corresponding experimental group, where LC terminals in the MPO/MnPO area had taken up the retro-AAV-DIO-caspase, body temperature of the mice was chronically lower. As was the case for the ΔLC mice, there were significant decreases in the maxima and minima of the body temperatures of retro-caspase-LC-lesioned mice recorded over 5 d compared with the control retro-caspase-LC-GFP mice, but the decrease in body temperature was confined to the “lights-off” period (SI Appendix, Fig. S2B). During “lights-off,” *retro-caspase–LC-*lesioned mice were about 1 °C cooler (average core body temperature) (SI Appendix, Fig. S2B), with decreases in both the highest and lowest body temperatures during “lights-off” compared with retro-LC-GFP controls (SI Appendix, Fig. S2B). Baseline sleep–wake profiles were not altered (SI Appendix, Fig. S2C). However, *retro-caspase–LC-*lesioned animals had a higher sensitivity to DEX (100 µg/kg): Compared with retro-caspase-LC-GFP controls, *retro-caspase–LC-*lesioned mice stayed in NREM-like sleep longer (probably because of hypothermia), and experienced a slower recovery from hypothermia (SI Appendix, Fig. S2D). These effects were similar to the increased sensitivity of ΔLC mice to DEX (Fig. 2D).
DEX Induces Hypothermia via adra2a Receptors Expressed on Glutamate Neurons in the Medial PO Hypothalamus.
The LC has a strong expression of adra2a receptors (Fig. 1B). But having found the LC and its associated adra2a receptors are dispensable for DEX-induced sleep and hypothermia, we looked for relevant DEX targets (i.e., adra2a receptors) in the forebrain. There, adra2a gene expression occurs mainly in layer VI cortical pyramidal cells, lateral septum (LS) and MPO hypothalamus (SI Appendix, Fig. S2E) (17, 53). The enhanced sensitivity of LC-lesioned (ΔLC) mice to DEX could arise from upregulation of adra2a receptors because of a reduced NE input from the LC. Indeed, adra2a mRNA levels in ΔLC mice were increased about twofold in both medial PO hypothalamus and LS (SI Appendix, Fig. S2F).
We previously generated and tested a knockdown adra2a shRNA that blocked the actions of DEX on LC cells (18). We used the same adra2a shRNA, modified into an AAV transgene that was Cre recombinase-dependent, to knock down adra2a gene expression selectively in glutamate and GABA neurons of the LS and MPO hypothalamic area of VGlut2-Cre and Vgat-Cre mice (that had nonlesioned LCs). AAV-DIO-shRNA-adra2a and AAV-DIO-shRNA-scramble (scr), each mixed with AAV-DIO-mCherry (to mark the sites of shRNA transgene expression), were injected bilaterally into the LS and M (medial) PO hypothalamus (each injection covered both the MPO and septal sites) of Vgat-Cre and Vglut2-Cre mice to generate Δadra2a-MPO/septum-Vglut2 and Δadra2a-MPO/septum-Vgat mice, respectively. The amounts and distribution of baseline sleep of Δadra2a-MPO/septum-Vgat and Δadra2a-MPO/septum-Vglut2 mice was unchanged from scr shRNA controls, and there was no difference in core body temperature compared with controls (SI Appendix, Fig. S3 A and B).
In the Δadra2a-MPO/septum-Vgat mice (SI Appendix, Fig. S4A), adra2a shRNA transgene expression was strong in both the LS and MPO areas, reflecting the expression of the endogenous vgat gene (SI Appendix, Fig. S4A). Δadra2a-MPO/septum-Vgat and scr control mice (ambient temp was 21 °C) were given 25, 50, and 100 µg/kg DEX. At all doses, there was no difference between mouse groups in DEX’s ability to induce NREM-like sleep or hypothermia, and recovery time was also unchanged (SI Appendix, Figs. S3C and S4C). Thus, GABA neurons in the MPO area do not contribute to adra2a receptor agonist actions for hypothermia or NREM-like sleep.
In Δadra2a-MPO/septum-Vglut2 mice, adra2a shRNA transgene expression was mainly restricted to the MPO hypothalamus and the septal hypothalamic nucleus (there are few endogenous Vglut2-expressing cells in the LS, hence little AAV transgene expression there) (Fig. 5A). Scr and shRNA mouse groups were given 25, 50, and 100 µg/kg doses of DEX. At 100 µg/kg, there was no difference between the groups (SI Appendix, Fig. S3C), but the lower doses of 25 and 50 µg/kg DEX were less effective in maintaining NREM-like sleep in Δadra2a-MPO/septum-Vglut2 mice, although both doses induced sleep equally well as mice treated with scr shRNA controls (Fig. 5B). There was, with both doses, the same amount of induced NREM-like sleep as found in scr mice, but within 30 min Δadra2a-MPO/septum-Vglut2 mice had started to wake up again compared with scr controls (Fig. 5B). By 8 h post–DEX injection, the wakefulness of Δadra2a-MPO/septum-Vglut2 mice injected with DEX were already similar to saline-injected controls. By contrast, the scr control mice injected with DEX were still sedated at 8 h postinjection.
*DEX induces hypothermia via adra2a receptors expressed on glutamatergic neurons in the MPO hypothalamus. (A) Left-hand panel: Schematic of knockdown of adra2a receptor transcripts in the MPO area of Vglut-2-Cre mice. Middle panel: expression of AAV-adra2a-shRNA transgene in the medial preoptic area (MPO), LS, and septo-hypothalamic (SHy) nucleus, as detected by immunocytochemical staining for mCherry (coronal section). Right-hand panel: Heatmap documenting the pooled distribution of AAV-adra2a-shRNA transgene expression in n = 6 mice, as detected by mCherry staining. Color key indicates the number of mice that had transgene expression in the area. CPu, Caudate putamen; Ctx, Cortex. (Scale bar, 1,000 µm.) (B) DEX-induced NREM-like sleep maintenance is diminished by knocking down adra2a receptor expression in MPO glutamatergic cells. Amounts of wake, NREM-like, and REM sleep over time following saline or ascending doses of DEX injections in Δadra2a-MPO/septum-Vglut-2 (n = 5) and controls (scr) (n = 5) mice (two-way ANOVA, *P < 0.05, **P < 0.01, ****P < 0.001). Shading indicates SEM. Mice were at ambient temperature. (C) DEX-induced hypothermia produced with 25 µg/kg and 50 µg/kg DEX requires adra2a receptors on MPO glutamatergic neurons. Curves for temperature and time following DEX i.p. injections in Δadra2a-MPO/septum-Vglut-2 (n = 5) and controls (scr) (n = 5) mice, two-way ANOVA, ****P < 0.001; graphs show the minimum body temperature (nadir) induced by each dose of DEX, which occurred at approx. 2 h after injection (nonpaired t test, ****P < 0.001 for 25 mg/kg DEX; and *P < 0.01 for 50 µg/kg DEX). Mice were at ambient temperature.
DEX at 25 and 50 µg/kg doses was less effective in inducing hypothermia in Δadra2a-MPO/septum-Vglut2 mice (Fig. 5C): The minimum temperature decrease was lower in the knockdown groups e.g., at 25 μg/kg, DEX caused an approx. minus 6 °C drop in body temperature, whereas in the control mice, the drop was around minus 11 °C (Fig. 5C). The recovery of body temperature was also faster in the Δadra2a-MPO/septum-Vglut2 mice compared with scr mice (Fig. 5C). Thus, a component of how DEX induces hypothermia resides in *adra2a-*expressing Vglut2 neurons in the MPO/septal hippocampal area.
Is the NREM-like sleep apparently evoked by adra2 receptors on MPO Vglut2 cells independent of temperature or a consequence of the hypothermia? To test this, following injection of DEX (25 and 50 µg/kg), Δadra2a-MPO/septum-Vglut2 and scr mice were given 2 h of external warming (SI Appendix, Fig. S5A). This external warming normalized the difference in sleep time for Δadra2a-MPO/septum-Vglut2 mice compared with scr controls that received the same DEX doses (SI Appendix, Fig. S5B). Thus, adra2a receptors on vglut2-expressing MPO neurons predominantly regulate DEX-induced hypothermia rather than NREM-like sleep.
Discussion
DEX, an important sedative used in intensive care, induces an arousable state in humans resembling (stages 2 and 3) NREM sleep (10??–13). A long-standing and popular theory is that DEX induces sedation by inhibiting the LC, a major source of NE (36??–39). We unexpectedly find that the LC helps maintain body temperature via NE/gal projections to the MPO hypothalamus. Without the LC, mice with no external warming become hypersensitive to DEX, causing deeper and longer hypothermia. This is likely because the key adra2a receptors regulating temperature and NREM sleep are not on the LC but elsewhere in the brain, including in the MPO hypothalamus (19, 54), and become upregulated in the absence of NE released from the LC (SI Appendix, Fig. S6). Without the LC, DEX has a greater effect on upregulated adra2a receptors to induce NREM-like sleep and hypothermia. When ΔLC mice are kept warm, DEX induces the same amount of NREM-like sleep as in mice with an intact LC, but with higher delta power. Our results show LC inhibition by DEX is not required for sedation. However, the LC speeds recovery from hypothermia. Thus, rather than being DEX’s prime target, the LC can be regarded instead as a “brake” on DEX’s hypothermic effects.
In agreement with earlier work where the LC was lesioned in rodents and cats (55, 56), we found LC lesioning does not affect baseline occurrence of sleep–wake states or the amount of sleep–wake fragmentation. Similarly, mice with selective deletion of the vesicular monoamine transporter 2 (vmat2) gene from all NE neurons (not just those in the LC), so that the cells cannot release NE, had no changes in the amounts of NREM, REM, and wake (57). We found that chemogenetically inhibiting the LC with CNO acting on hM4di receptors in LC neurons, mimicking DEX’s Gi inhibition, did not induce sleep or hypothermia, suggesting DEX likely does not act at the LC to induce NREM-like sleep. Similarly, opto-inhibiting LC cells does not cause sleep (58). The most distinct sleep-related trait in long-term LC-lesioned animals is their tendency to fall asleep more rapidly in unfamiliar environments (59). Giving ΔLC mice DEX, and providing external warming during and following the injections, induced much stronger NREM-like oscillations in the EEG compared with the delta amplitude when ΔLC mice at ambient temperatures were given DEX, further emphasizing that the LC is not needed for DEX-induced NREM-like sleep. Similarly, in rats with NE transmission removed by toxin depletion, DEX still lowered the halothane dose (minimum alveolar concentration) needed for anesthesia, suggesting DEX acts postsynaptically of the LC to induce sedation (40).
So, what is the LC doing with regard to DEX? In terms of general sleep–wake control, others have found that the LC aids waking up from stimuli: Optogenetically exciting LC neurons or their terminals in the PO hypothalamus induces waking from sleep (58, 60), and LC activity, by releasing NE into the thalamus during NREM sleep, aids arousability (61); chemogenetically exciting LC cells even induces arousal from general anesthesia (62); conversely, opto-inhibiting LC cells reduces the effectiveness of sounds to wake animals from NREM sleep (63). These observations are all consistent with the LC being required to reduce duration of DEX-induced NREM-like sleep. Chemogenetically inhibiting the LC prior to DEX administration substantially lengthened the duration of DEX-induced hypothermia and NREM-like sleep. As the effect of DEX is wearing off (i.e., DEX is metabolized), the LC is providing an arousal signal. An important part of this arousal effect is likely to be due to promoting recovery of normal body temperature.
Why are ΔLC mice more sensitive to DEX? Many types of cells upregulate receptors for a missing neurotransmitter. For example, denervating skeletal muscle elicits large increases in postsynaptic nicotinic acetylcholine receptor expression (64?–66). Similarly, in ΔLC mice, we speculate that the loss of NE tone causes the upregulation of adra2a receptors expressed on forebrain and hypothalamic neurons, making the associated circuits hypersensitive to the remaining NE input, including from other NE groups, such as the A1/C1 or A2/C2 brainstem groups that project to the forebrain (SI Appendix, Fig. S6). Because of the upregulated adra2a receptors on effector neurons in the LC-lesioned mice, DEX when given to these mice drives down temperature further and produces more hypothermia than in control mice. When mice without an LC are injected with DEX and kept warm for several hours, they have the same amount of NREM-like sleep as LC-GFP controls, although delta power is increased in the ΔLC mice, but this is less sleep than when the mice are sedated at ambient temperature (Fig. 3B). When the mice injected with DEX were unwarmed, the prolonged NREM-like sleep illustrated in Fig. 2D is likely caused by hypothermia. For example, perhaps clearance and degradation of the drug are delayed when the mice are colder.
DEX’s increased sedative and behavioral actions in ΔLC mice likely explain earlier observations on dopamine β-hydroxylase knockout mice (67). These mice, that develop from birth without the capacity to synthesize NE anywhere in the body, are behaviorally hypersensitive to DEX: They have about 67% shorter latencies to induce LORR, and anesthesia lasts up to 545% longer, suggesting an intact NE system is dispensable for DEX’s behavioral actions (67). (Temperature and NREM-like sleep were not measured in that study). From our results, the increased behavioral sensitivity of dopamine β-hydroxylase knockouts to DEX is likely due to upregulated adra2a expression.
Adra2a receptors are often inhibitory (68). In a part of the LPO, the ventral LPO hypothalamus, calcium imaging revealed that although systemically administered high doses of DEX (100 to 150 µg/kg, i.p.) inhibited many neurons, these doses also excited neurons that were active in NREM sleep (about 25% of GABA and around 6% of glutamate neurons) (69). Indeed, systemically injected DEX induces cFOS expression in the LS, bed nucleus stria terminalis (BNST), PO, and supraoptic hypothalamic nucleus (18, 41, 70). As cFOS induction requires neuronal excitation, this induction could be explained as originating by DEX inhibiting GABA interneurons that are, in turn, inhibiting the neurons that respond with cFOS induction (i.e., disinhibition). However, DEX can also excite neurons directly, as under some circumstances adra2a receptors couple to Gs proteins (68). DEX also directly depolarizes cells by reducing cAMP concentrations (via Gi) and thereby inhibiting hyperpolarization-activated cyclic nucleotide-gated cation channels which act as a clamp on the membrane potential, a mechanism that occurs in BNST neurons neighboring the PO area (71), and could also occur in the PO area too.
The PO hypothalamus is well known for housing circuitry that regulates sleep–wake states and temperature (49, 72). PO GABA cells can both promote and inhibit wake, depending on type and location (19, 21, 73, 74); similarly, PO glutamate cells can promote wake or sleep too, again depending on type and location (75??–78), and can induce hypothermia and torpor (77, 79); there is quite a range of the hypothermia-inducing glutamate cells in the PO, but these may actually converge to a few discrete subtypes of neurons which have common molecular markers (80).
What is a possible circuit mechanism for how DEX induces hypothermia? Chemogenetically activating gal cells in the LPO area induces NREM and drives down core body temperature substantially (19, 74), in a phenotype remarkably like the actions of DEX. On the other hand, lesioning gal LPO neurons reduces the effectiveness of DEX to induce NREM-like sleep and reduce body temperature (19), and the mice have permanently elevated core temperatures and strong-sleep–wake fragmentation (19), implying gal LPO cells are part of the “NREM sleep-on” and “temperature decrease” switches. The gal LPO cells are likely GABAergic, but a subset could use glutamate (81). Yet adra2a receptors on GABA (vgat) cells in MPO are not responsible for DEX-induced sleep and hypothermia. Rather, only knockdown of adra2a receptors on MPO glutamate (Vglut2) cells blunted hypothermia induction, and did not affect sleep induction, and when the animals were externally warmed, DEX-induced NREM-like sleep was unaffected by adra2a knockdown on MPO glutamate cells. Thus, the simplest hypothesis is DEX binds to excitatory adra2a receptors on MPO glutamate cells, which then excite a subset of LPO gal cells to drive down temperature and induce NREM-like sleep (SI Appendix, Fig. S6). Also, in LPO, DEX would activate NREM sleep-inducing gal cells, but these might be a different subpopulation from the ones driving hypothermia. It could also be that the actual induction of NREM-like sleep (delta waves in the EEG) by DEX requires the adra2a receptors expressed on layer VI pyramidal neurons (SI Appendix, Fig. S2E), as well as neurons in the supraoptic area (SON). The supraoptic area (SON) in the hypothalamus and neurons in the suprachiasmatic (SCN) nucleus may also contribute to NREM-like sleep induction by DEX (41, 42), though these studies (both using 100 µg/kg DEX) did not document body temperature. SON neurons identified by cFOS trapping regulated anesthetic responses from various agents—not just DEX—and natural NREM sleep induction (41). How, and if, these SON and SCN neurons regulate temperature is unclear.
At the highest DEX dose (200 μg/kg, i.p.) in ΔLC mice, DEX caused 100% mortality in unwarmed mice—they did not recover from extended hypothermia; but when kept warm, the same dose was tolerated (Fig. 3 A and B). The cause of death is unknown, but hypothermia and sustained bradycardia are probable factors, probably due to overstimulation of adra2a receptors. DEX clearance from the brain and body will also be slower during hypothermia, causing the drug’s effects to persist. What about off-target effects? At 100 μg/kg DEX, α2a knockout mice showed no hypothermic response; at 300 μg/kg (higher than our dose), they had marginal responses; and even 1,000 μg/kg DEX produced only a small hypothermic effect (23). Thus, off-target effects of the maximal DEX dose we used are possible, but less likely.
When DEX is given in the clinic, strenuous efforts are made to keep patients warm, but there is, however, still a risk of inadvertent hypothermia and long-term pathological consequences. Hypothermia is a double-edged sword (27). Controlled and induced hypothermia can protect organs (27). On the other hand, there are many complications of hypothermia (26, 28, 29): For example, a two-degree decrease in core body temperature during a surgical operation triples the risk of wound infections (26), and may trigger postoperative cognitive deficits. These could arise because cooler temperatures during anesthesia slow turnover of phosphorylated forms of the microtubule binding protein tau (the phosphatases work less well), potentially seeding neurodegenerative disease (30). Even in healthy humans, a chronically lower core body temperature is associated with increased phosphorylated tau (82). In this respect, based on the results with ΔLC mice, a fully functioning LC likely protects against adverse outcomes of anesthesia. Even when ΔLC mice were in a warm environment, although the extra hypothermia following DEX administration was lessened, there was still more than in control mice with an intact LC. In Alzheimer’s disease (AD), LC neurons are some of the first to accumulate hyperphosphorylated tau, and up to 80% of LC neurons die eventually (83), which is about the same amount achieved by the genetic LC lesions in our study. This suggests that, because of premature LC damage in people in the early phases of AD, some years before they manifest cognitive deficits, they would be at slightly more risk during general anesthesia.
There are some caveats to our results: The lack of hypothermia in patients sedated with DEX may be attributed, as shown in our experiments, to the presence of an intact LC, or to hypothermia being dependent on environmental temperature. Alternatively, it may be because the clinical doses of DEX are significantly lower than those used in animal studies. For example, in experimental studies, humans are typically infused with doses of DEX in the range 1 to 6 µg/kg/h (24, 84, 85), and in clinical practice, infusion rates of 0.7 to 1.4 µg/kg/h are recommended (see data in refs. 3 and 85). Nevertheless, human volunteers given 2 µg/kg DEX experienced about a one degree drop in their body temperature (24).
Although hypothermia may not be a significant side effect of DEX in clinical practice, our study provides insight into the mechanism by which the drug, by removing temperature control, lowers the temperature threshold for initiating vasoconstriction and shivering in humans (31). Thus, DEX can be used to reduce shivering when given after anesthesia (31, 33, 86). This property of DEX could have practical clinical applications, such as reducing postoperative shivering after general anesthesia (33), providing sedation for neonates with hypoxic-ischemic encephalopathy undergoing therapeutic hypothermia (87), or for sedating cardiac arrest patients receiving targeted temperature management.
In summary, we have shown that the key textbook mechanism for how α2 adrenergic sedatives are supposed to induce NREM-like sleep and hypothermia by inhibiting the LC is unlikely. The hypothermia actions of DEX are mediated in part by adra2a receptors on PO hypothalamic glutamate (Vglut2) neurons. On the other hand, the LC does promote recovery from hypothermia and is likely to be protective from any adverse effects of hypothermia.
Materials and Methods
Full details of transgene construction, AAV production, stereotaxic injection coordinates, drug concentrations, EEG and temperature recording, sleep scoring, chemogenetics, histology and immunostaining, RNAscope (Advanced Cell Diagnostics), and statistics are given in SI Appendix. Experiments were in accordance with the United Kingdom Home Office Animal Procedures Act (1986) and were approved by Imperial College’s Animal Welfare and Ethical Review Body. Adult mice of both sexes were used. Gal-Cre mice were a gift from N. Heintz (The Rockefeller University, NY) and obtained from the Mutant Mouse Regional Resource Center (stock #031060-UCD) (88); Vgat-Cre mice (89) and Vglut-2 Cre mice (89) were a gift from B. B. Lowell (Harvard Medical School, Boston) and obtained from the Jackson Laboratory (JAX stock #016963 and #016962, respectively). All AAVs (mixed serotype 1/2 and retrocaspase) were produced in-house as described previously (90). The rAAV2 packaging plasmid was a gift from Alla Karpova (Janelia Research Campus, VA) and David Schaffer (University of California, Berkeley, CA) (Addgene plasmid #81070) (52); pAAV-DIO-GFP was a gift from John T. Gray (University of California Davis, CA) (Addgene plasmid 32396); pAAV-EF1α-DIO-taCASP3-TEV was a gift from Nirao Shah (Stanford University, CA) (Addgene plasmid 45580) (91); pAAV-DIO-hM4Di-mCherry was a gift from Bryan L. Roth (University of North Carolina, NC) (Addgene plasmid 44362) (92). For constructing the Cre recombinase-dependent adra2a shRNA knockdown and scr shRNA transgenes, we used the pPRIME system based on an mir30 expression cassette (93). EEG recordings were made with Neurologger 2A devices (94). Temperature loggers (DST nano, Star-Oddi) were intra-abdominally implanted as described previously (77).
Supplementary Material
Appendix 01 (PDF)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1N. P. Franks, W. Wisden, The inescapable drive to sleep: Overlapping mechanisms of sleep and sedation. Science 374, 556–559 (2021).34709918 10.1126/science.abi 8372 · doi ↗ · pubmed ↗
- 2H. Scheinin, R. Virtanen, E. Mac Donald, R. Lammintausta, M. Scheinin, Medetomidine–A novel alpha 2-adrenoceptor agonist: A review of its pharmacodynamic effects. Prog. Neuropsychopharmacol. Biol. Psychiatry 13, 635–651 (1989).2571177 10.1016/0278-5846(89)90051-1 · doi ↗ · pubmed ↗
- 3M. A. S. Weerink , Clinical pharmacokinetics and pharmacodynamics of dexmedetomidine. Clin. Pharmacokinet. 56, 893–913 (2017).28105598 10.1007/s 40262-017-0507-7PMC 5511603 · doi ↗ · pubmed ↗
- 4K. T. Ng, C. J. Shubash, J. S. Chong, The effect of dexmedetomidine on delirium and agitation in patients in intensive care: Systematic review and meta-analysis with trial sequential analysis. Anaesthesia 74, 380–392 (2019).30367689 10.1111/anae.14472 · doi ↗ · pubmed ↗
- 5Y. Skrobik , A randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 197, 1147–1156 (2018).29498534 10.1164/rccm.201710-1995 OC · doi ↗ · pubmed ↗
- 6X. Yu, N. P. Franks, W. Wisden, Sleep and sedative states induced by targeting the histamine and noradrenergic systems. Front. Neural Circuits 12, 4 (2018).29434539 10.3389/fncir.2018.00004 PMC 5790777 · doi ↗ · pubmed ↗
- 7G. M. Keating, Dexmedetomidine: A review of its use for sedation in the intensive care setting. Drugs 75, 1119–1130 (2015).26063213 10.1007/s 40265-015-0419-5 · doi ↗ · pubmed ↗
- 8L. Sattar , Comparison between Dexmedetomidine and Propofol for sedation on outcomes after cardiac surgery in patients requiring mechanical ventilation: A meta-analysis of randomized-control trials. Cureus 15, e 42212 (2023).37609090 10.7759/cureus.42212 PMC 10441820 · doi ↗ · pubmed ↗