The pathophysiological mechanisms of immunosenescence in coronary artery disease
Hengjie Bie, Zhengxian Tao

TL;DR
This paper explores how aging-related immune decline, called immunosenescence, contributes to coronary artery disease and discusses potential treatments.
Contribution
The paper highlights recent advances in intervention strategies targeting immunosenescence in coronary artery disease.
Findings
Immunosenescence promotes chronic inflammation and atherosclerosis through immune cell dysfunction.
Interventions like thymic regeneration and macrophage modulation show promise in treating CAD.
Future research should focus on personalized therapies and clinical validation.
Abstract
Coronary artery disease (CAD) is the most common coronary heart disease, characterized by the accumulation of atherosclerotic plaques in the coronary arteries, which supply oxygen and nutrients to the heart. The National Health and Nutrition Examination Survey (NHANES) reported that between 2011 and 2014, the prevalence of coronary artery disease was higher in men (30.6%) than in women (21.7%) aged ≥80 years. In the ARIC (Atherosclerosis Risk in Communities) study, the incidence of myocardial infarction (MI) was higher in black individuals compared to white individuals among those aged 65–84 years. Immunosenescence plays a pivotal role in its onset and progression. Immunosenescence is a complex process involving organ remodeling and cellular regulation, leading to a decline in immune function and reduced responses to infection and vaccination in older adults. By driving dysfunction in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
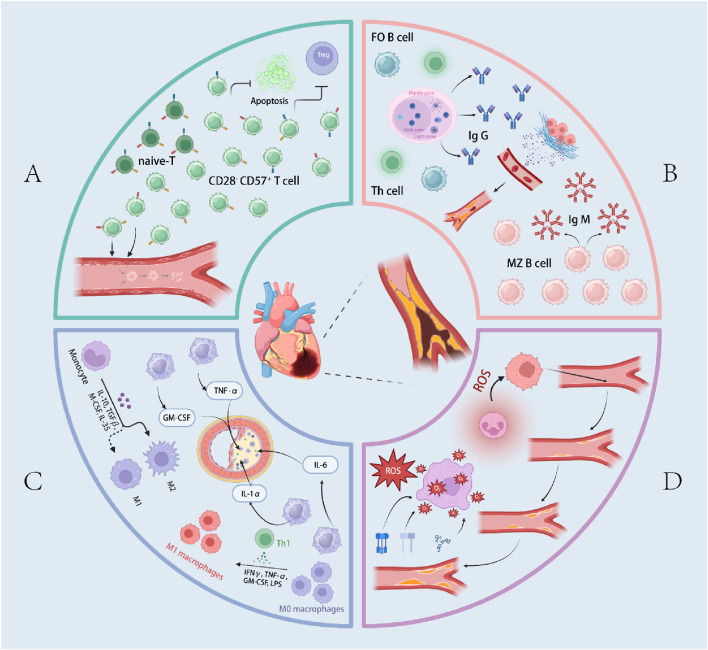 FIGURE 1
FIGURE 1| Module | Aging shift | CAD effect | Key interventions (ultra-brief) |
|---|---|---|---|
| T cells | Naïve ↓; CD28−CD57+ ↑; TCR diversity ↓ | Endothelial injury; inflammation/ROS ↑ → atherogenesis | IL-7 axis; CMV control + precision vaccines; sestrin–p38 block; selective depletion (e.g., CD153); cautious telomere support |
| B cells | FO IgG to OSEs ↑; MZ/B1 IgM ↓ | IgG fuels plaques; IgM clears oxLDL/apoptotic cells | Deplete ABCs/AABs; rebuild B-1/natural IgM; IL-1RA or anti-CD20 |
| Macrophages | Phagocytosis/TLR/MHC-II ↓; M1/M2 imbalance; SASP/ROS ↑ | Chronic inflammation; poor resolution; thicker neointima | IL-2/anti-CD40 reprogramming; CR mimetics (metformin/resveratrol/rapamycin); oral p38 inhibition; boost TLR/MHC-II |
| Inflammaging | Persistent cytokines; DAMP–PRR; ROS/RNS ↑; CMV imprint | Endothelial damage; VSMC proliferation; foam cells; unstable plaques | CR/antioxidants; senolytics; vaccine/pathogen management |
| HSCs | DNA damage/ROS; telomere attrition; cdc42 ↑ | Myeloid bias; lymphoid output ↓ | p38 + antioxidants; Sirt3/autophagy (CMA) restore; telomere maintenance*; cdc42 inhibition (CASIN); young-donor grafts |
| System-level | Thymic involution; metabolic/epigenetic drift; senescent burden ↑ | Immune regeneration ↓; inflammaging ↑ | FOXN1-based thymic regeneration; CR/metformin/rapamycin/spermidine/α-KG; senolytics (β-gal prodrugs, uPAR CAR-T); SIRT6/DNMT/HDAC modulation |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAtherosclerosis and Cardiovascular Diseases · Neutrophil, Myeloperoxidase and Oxidative Mechanisms · Adipokines, Inflammation, and Metabolic Diseases
1 Introduction
Coronary artery disease (CAD) is the most common type of coronary heart disease, characterized by the accumulation of atherosclerotic plaques in the coronary arteries, which supply oxygen and nutrients to the heart (Tsao et al., 2022). It is a leading cause of mortality and disability, with approximately 380,000 deaths in 2020. The NHANES reported a higher prevalence of CAD in men (30.6%) than in women (21.7%) aged ≥80 years between 2011 and 2014 (Benjamin et al., 2017; National Heart and Institute, 2006; Global, 2015). Similar findings were observed in the FHS (Framingham Heart Study) and CHS (Cardiovascular Health Study). In the ARIC (Atherosclerosis Risk in Communities) study, the incidence of myocardial infarction (MI) was higher in black individuals compared to white individuals among those aged 65–84 years. CAD is a chronic inflammatory disorder, with atherosclerosis as the central pathological process. Clinically, it presents as stable angina, unstable angina, myocardial infarction (MI), or sudden cardiac death (Ross, 1999; Álvarez-Álvarez et al., 2017). Atherosclerosis involves the formation of plaques in the vascular intima, primarily driven by chronic inflammation due to various risk factors, including cholesterol accumulation and the retention of lipoproteins such as low-density lipoprotein (LDL) in the arterial walls (Basatemur et al., 2019; Soehnlein and Libby, 2021).
The assessment of CAD can be categorized into anatomical and functional approaches (de Oliveira Laterza Ribeiro et al., 2023). Anatomical evaluation is primarily performed using coronary angiography and coronary computed tomography (CCT), the latter offering advantages in noninvasive imaging, vascular structural analysis, and prediction through coronary artery calcium (CAC) scoring. However, anatomical methods have limitations in determining the functional significance of coronary lesions; therefore, functional assessment serves as an essential complement. Commonly used functional techniques include exercise electrocardiography (ECG), stress echocardiography, single-photon emission computed tomography (SPECT), cardiac magnetic resonance imaging (CMR), and positron emission tomography (PET), with these imaging modalities demonstrating diagnostic accuracies exceeding 80% (Takx et al., 2015). The primary objectives of CAD treatment are to alleviate symptoms and prevent complications in patients with stable CAD, while for those with acute coronary syndrome (ACS), the goal is to improve coronary blood flow and restore cardiac function as rapidly and effectively as possible (Ford et al., 2018; Herrmann et al., 2012). Traditionally, these goals are achieved through pharmacological therapy, with consideration of percutaneous coronary intervention (PCI) or coronary artery bypass grafting (CABG) when necessary. For patients with stable CAD, pharmacological management typically includes beta-blockers, calcium channel blockers, nitrates, angiotensin-converting enzyme inhibitors (ACEIs), and statins, whereas for patients with ACS, treatment primarily involves thrombolytic agents and antiplatelet therapies (Kh et al., 2017). In recent years, beyond traditional risk factors (Yusuf et al., 2004)—including hyperlipidemia, hypertension, diabetes mellitus, and cigarette smoking—alterations in immune system function (Jonasson et al., 2003), particularly immunosenescence (Santoro et al., 2021; Liu et al., 2023), have been recognized as critical drivers of the onset and progression of CAD.
Immunosenescence is a complex process involving organ remodeling and multilayered cellular regulation, characterized by a progressive decline in immune competence that compromises responses to infections and vaccinations in older adults (Nikolich-Žugich, 2018; Lanna et al., 2017; Ucar et al., 2017). Key features include thymic involution, hematopoietic stem cell (HSC) dysfunction, altered naïve-to-memory T- and B-cell ratios, chronic low-grade inflammation (“inflammaging”), senescent cell accumulation, impaired recognition of novel antigens, mitochondrial dysfunction, metabolic dysregulation, and increased genomic instability. With advancing thymic atrophy (Goronzy and Weyand, 2013), functional epithelial regions are gradually replaced by non-thymopoietic perivascular spaces, resulting in reduced peripheral naïve T cells, expansion of terminally differentiated memory T cells, and restricted T-cell egress.
Adults thymectomized in early childhood exhibit premature immunosenescent profiles. Nevertheless, thymic degeneration alone does not fully account for diminished T-cell receptor (TCR) diversity, and thymic rejuvenation may not restore it. Immunosenescence arises from the persistent accumulation of endogenous damage, where senescent cells sustain low-grade chronic inflammation through the secretion of a senescence-associated secretory phenotype (SASP), which includes IL-1, IL-6, IL-8, IL-13, IL-18, TNF, and their corresponding receptors. Cellular senescence exemplifies pleiotropy—providing early-life benefits by suppressing tumorigenesis and promoting tissue development and repair, but exerting detrimental effects in later life (Feldman et al., 2015). Metabolic reprogramming accompanying immunosenescence—characterized by increased glycolysis, mitochondrial dysfunction, and elevated reactive oxygen species—closely associates with the high late-life incidence of cardiovascular, neurodegenerative, autoimmune, and metabolic diseases, as well as cancer. This review summarizes the mechanistic roles of immunosenescence in CAD and discusses its therapeutic potential, with the aim of informing new strategies for the prevention and treatment of CAD.
2 Immunosenescence and CAD
2.1 T cell
As the immune system ages, the balance of immune cell populations shifts, characterized by a decline in naïve T cells and an expansion of memory and senescent T cells (Nikolich-Žugich, 2018; Ghamar Talepoor and Doroudchi, 2022). Senescent T cells typically lose CD28 and acquire CD57 expression; these cells are terminally differentiated, exhibit potent pro-inflammatory and cytotoxic activity, and are resistant to apoptosis and regulation by regulatory T cells (Tregs) (Vallejo et al., 1999; Formentini et al., 2021; Álvarez-Heredia et al., 2023; Broadley et al., 2017). In addition to reductions in proliferative naïve T and B cells with a relative increase in central memory subsets, aging is accompanied by contraction of the T-cell receptor (TCR) repertoire, resulting in diminished antigen responsiveness. Studies have shown that CD28^−^CD57^+^ T cells can directly or indirectly kill endothelial cells (Namekawa et al., 2000), thereby contributing to vascular injury. Expansion of CD28^−^CD57^+^ T cells is associated with heightened inflammatory responses and increased oxidative stress, both of which promote atherogenesis and the pathogenesis of CAD (Téo et al., 2013; Dumitriu et al., 2009). Consequently, this phenotypic shift in T cells, together with other age-related immune alterations, sustains a chronic inflammatory milieu that drives the initiation and progression of CAD (Bergström et al., 2012).
2.2 B cell
B cells exert bidirectional regulation in atherosclerosis. B2 cells constitute the predominant B-cell pool in the circulation and secondary lymphoid organs; among them, follicular B cells (FO B cells) are pro-atherogenic (Snijckers and Foks, 2024). Through cooperation with T follicular helper (Tfh) cells, FO B cells undergo activation and enter germinal centers, differentiating into GC B cells that produce high-affinity IgG (Sage et al., 2019) autoantibodies against oxidation-specific epitopes (OSEs)—including stress-induced endothelial HSP60/65, malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) generated from oxLDL, and phosphocholine-containing oxidized phospholipids. These IgG antibodies increase under hyperlipidemic conditions, amplifying inflammation and enlarging plaques. Experimentally, engagement of the BTLA pathway reduces FO B cells by ∼50% and significantly decreases plaque burden; FO deficiency induced by targeted deletion of Blimp-1 or Pax5 similarly lowers IgG levels and attenuates lesions. Conversely, transfer of IgG from atherosclerotic mice into Ldlr−/− mice lacking endogenous IgG aggravates plaque development. In contrast to FO B cells, marginal zone (MZ) B cells suppress the atherogenic functions of Tfh cells and, in a T-cell–dependent manner, produce atheroprotective IgM. B1a/B1b cells likewise confer protection by secreting large amounts of germline-encoded natural IgM recognizing OSEs, thereby promoting non-immunogenic clearance of oxLDL and apoptotic cells (Centa et al., 2018; Binder et al., 2016). IL-10^+^ regulatory B cells inversely correlate with plaque severity, although (Tsiantoulas et al., 2012) B-cell–specific IL-10 deficiency does not alter lesion size, suggesting that their protective effects may be partly—or largely—independent of IL-10 (Douna et al., 2019).Overall, the impact of B cells on atherogenesis is determined by subset identity, crosstalk with Tfh cells, and antibody isotype: IgG generally promotes plaque progression, whereas IgM primarily mediates neutralization and clearance (Sage et al., 2015).
2.3 Macrophage
The changes in macrophages during immunosenescence represent a complex process driven by multiple factors, centered on the interplay between functional impairment and the acquisition of cellular senescence-associated characteristics (Sharma, 2021). Macrophages span both the initiation and the resolution phases of inflammation, and their M1 (pro-inflammatory) versus M2 (pro-repair) polarization balance shifts with age in a tissue- and context-dependent manner: a bias toward M2 has been reported in aging skeletal muscle and in macular degeneration, whereas a shift toward M1 has been attributed to enteric nervous system degeneration in aged mice (Mahbub et al., 2012; Wynn et al., 2013; Cui et al., 2019; Zandi et al., 2015; Sharma et al., 2014). Regardless of direction, any imbalance disrupts host defense and resolution programs, rendering older individuals more susceptible to infections and chronic inflammation. Functionally, aged macrophages display reduced phagocytosis and respiratory burst (Chelvarajan et al., 2006; Linehan et al., 2014; Wong et al., 2017; Swift et al., 2001), diminished TLR expression (Davila et al., 1990; Herrero et al., 2001), blunted responses to antigenic stimulation, and lowered LPS-induced cytokine production (Ding et al., 1994). Notably, bone-marrow–derived macrophages do not fully recapitulate these defects, indicating that age-associated dysfunction is not purely cell-intrinsic but is strongly shaped by extrinsic microenvironmental cues. Antigen presentation is likewise compromised: after IFN-γ stimulation, induction of MHC class II and the capacity to activate T cells are reduced, slowing and weakening adaptive immunity. In tissue repair, aging alters chemokine milieus and patterns of macrophage infiltration, delays efferocytosis, and thereby impairs wound healing. Following vascular injury, aged arteries form thicker neointimas, correlating with increased tissue macrophages and elevated IL-18; depletion of macrophages mitigates this phenotype.
From the perspective of cellular senescence, macrophages in old age or in DNA-repair–deficient settings upregulate p16INK4a and p21CIP1, acquire a SASP, adopt elongated morphologies, and exhibit SA-β-gal activity—features consistent with their serving as in vivo sources of SASP that amplify inflammation. That said, some evidence suggests p16/SA-β-gal expression in macrophages can reflect reversible activation states rather than bona fide senescence, underscoring the need to distinguish these conditions rigorously. Telomere biology provides a more direct etiologic thread: age-related telomere attrition or telomerase loss drives oxidative stress, mitochondrial abnormalities, and hyperactivation of the NLRP3 inflammasome, thereby impairing phagocytosis, GM-CSF–dependent proliferation, and signaling (e.g., Stat5a oxidation/phosphorylation). These changes align with inflammatory phenotypes observed in chronic diseases such as type 2 diabetes and sickle cell disease. Accumulating ROS both drives senescence and reinforces immune dysfunction, creating a feed-forward loop (Frodermann and Nahrendorf, 2018; Frangogiannis, 2021).
In summary, macrophage immunosenescence is sculpted by intertwined intrinsic factors (telomere/mitochondrial damage, cell-cycle inhibition, SASP) and extrinsic factors (aging tissue microenvironments and low-grade sterile inflammation). The result is a coexistence of effector hypofunction with dysregulated inflammatory control. Although definitive causal chains linking cellular senescence and immunosenescence in macrophages remain to be established, current evidence nominates actionable targets focused on reducing oxidative stress, rebalancing M1/M2 polarization, restoring antigen presentation, and constraining the SASP.
2.4 Inflammaging
“Inflammaging” refers to the chronic, low-grade inflammatory state that emerges during aging (Franceschi et al., 2018). It is primarily macrophage-centered and characterized by a complex balance between pro-inflammatory and anti-inflammatory responses. According to current literature, the major sources of inflammatory stimuli are endogenous, mislocalized, or structurally altered molecules derived from damaged or dying cells and organelles (cellular debris), which are recognized by receptors of the innate immune system. Although the production of these molecules is physiological and increases with age, their clearance through autophagy, mitophagy, and proteasomal degradation progressively declines. This “autoreactive/autoimmune” process drives the onset and progression of chronic diseases and can accelerate and amplify aging both locally and systemically.
“Inflammaging” is a major risk factor for cardiovascular disease (CVD), characterized by persistently elevated pro-inflammatory cytokines that drive endothelial injury, impaired vascular remodeling, and atherosclerosis (Franceschi et al., 2018; Donato et al., 2015). These cytokines arise predominantly from senescent T cells and pro-inflammatory macrophages (Libby et al., 2009), reflecting age-related failures in immune regulation that disrupt tissue homeostasis (Leon and Zuckerman, 2005; Franceschi and Campisi, 2014). During cardiac stress, ischemic injury, or metabolic syndrome, necrotic cells release damage-associated molecular patterns (DAMPs) that are sensed by pattern-recognition receptors (PRRs) on innate immune cells, triggering robust inflammation (Mauro et al., 2019). The ensuing secretion of pro-atherogenic cytokines, reactive oxygen species (ROS), and reactive nitrogen species (RNS) amplifies oxidative stress, promotes vascular smooth muscle cell (VSMC) proliferation, and fosters the accumulation of oxidized low-density lipoprotein (LDL), which is taken up by macrophages to form foam cells and destabilize plaques. On the T-cell axis, expansion of senescence-associated cytotoxic populations—particularly CD8^+^CD28^−^ T cells—is closely linked to vascular dysfunction, while late-differentiated CD4^+^CD28^−^ T cells that produce IFN-γ increase in unstable angina. At the population level, cytomegalovirus (CMV)–associated atherosclerosis in older men may be mediated by a higher proportion of memory CD4^+^ T cells. Collectively, an aging immune system, through DAMP–PRR–driven inflammatory amplification and T-cell repertoire remodeling, orchestrates endothelial damage, oxidative stress, lipid deposition, and maladaptive vascular remodeling, forming the immunologic substrate for CVD onset and progression. The above-mentioned pathogenesis is shown in Figure 1.
Overview of immunosenescence in Coronary artery disease (CAD). (A) represents the pathogenic mechanisms of T cells in CAD within the context of immunosenescence. (B) illustrates the pathogenic mechanisms of B cells in CAD within the context of immunosenescence. (C) depicts the pathogenic mechanisms of macrophages in CAD within the context of immunosenescence. (D) outlines the pathogenic mechanisms of inflammaging in CAD. The terms “follicular B cells (FO B cells)”, “marginal zone B cells (MZ B cells)”, and “reactive oxygen species (ROS” are also included.
3 Therapeutic strategy
3.1 Immune progenitors (hematopoietic stem cells)
Anti-aging interventions for hematopoietic stem cells (HSCs) can be implemented as an integrated program of “damage mitigation, metabolic stabilization, telomere protection, polarity correction, and source replacement.” First, to counter DNA damage and elevated reactive oxygen species (ROS) (Brown et al., 2013) in aged HSCs, inhibition of the p38 stress pathway (Hsieh and Papaconstantinou, 2002; Li et al., 2011) together with enhancement of antioxidant defenses can lower oxidative stress and improve self-renewal and hematopoietic potential; in parallel, restoring mitochondrial homeostasis (e.g., via upregulation of Sirt3) and re-establishing autophagy—particularly reactivation of chaperone-mediated autophagy (Dong et al., 2021) (CMA)—helps correct energetic imbalance and bolster hematopoietic resilience. Second, preservation of telomere integrity—within strict safety boundaries—through telomerase activation/gene therapy or reinforcement of telomere-binding proteins may alleviate telomere-driven DNA damage responses and myeloid skewing (Allsopp et al., 2003), while necessitating careful stratification given potential oncogenicity. Third, correcting cell polarity and lineage bias by inhibiting hyperactive cdc42 (Hosokawa et al., 2017; Hammad and Lambrecht, 2008) (e.g., with the small molecule CASIN) can restore HSC polarity, enhance lymphoid progenitor output, and re-establish immune homeostasis, with evidence for broader rejuvenation across stem-cell compartments. Fourth (Kovina et al., 2019), “source replacement” via transplantation of bone marrow/hematopoietic grafts from young donors (Das et al., 2019) extends lifespan and improves neurobehavior in animal models, and in humans the donor’s age imprints the epigenetic age of reconstituted blood, suggesting a feasible route to immune system reset in older individuals. Overall, a personalized combination of p38/oxidative control, metabolic and autophagy correction, telomere maintenance, cdc42 inhibition, and—when appropriate—transplantation is recommended, coupled with rigorous oncologic surveillance and long-term follow-up.
3.2 T and B cell
Interventions for T-cell aging in older adults can be advanced through an integrated framework of “rebuilding numbers, preserving diversity, restoring function, and selectively de-senescing.” First, reconstituting the IL-7/IL-7R axis to expand the peripheral T-cell pool is supported by clinical data in HIV cohorts showing recombinant human IL-7 increases and activates both CD4^+^ and CD8^+^ T cells, providing a rationale for physiological recalibration in healthy elders. Second, limiting TCR repertoire contraction and clonal expansions—via pathogen management (e.g., CMV) and optimized, precision vaccination—helps maintain the naïve pool and antigen-recognition breadth. Third, reversing maladaptive phenotypic remodeling by inhibiting the sestrin–p38 stress pathway can prevent the age-associated shift of CD8^+^ T cells from TCR-dependent to NK-like activity, thereby restoring antigen specificity and enhancing vaccine responsiveness. Fourth, phenotype-guided, selective removal or reprogramming of “senescence-like” subsets (Yoshida et al., 2020) (CD28 low/−, CD27^−^, CD57^+^, CD45RA^+^ CD27^−^) is feasible; notably, a CD153-targeted peptide vaccine safely reduces CD153^+^ senescent T cells in mice and improves metabolic homeostasis, illustrating precise depletion without compromising normal immunity. Fifth (Lanna et al., 2017; Martínez and Blasco, 2017),correcting cellular senescence at its roots through telomere/telomerase-directed strategies—including indirect telomerase upregulation via the sestrin–p38 axis—may mitigate DNA damage and pro-inflammatory phenotypes while restoring proliferative capacity and durability. Overall, stratification by immune phenotype and combination regimens that couple IL-7 supplementation (Fry and Mackall, 2005; Rosenberg et al., 2006), sestrin–p38 inhibition, telomere maintenance, and targeted vaccination are recommended, with continuous monitoring for infection risk, hyperactivation or autoimmunity, and metabolic adverse effects to maximize the quantity, quality, and specificity of adaptive immunity in older individuals.
Therapeutic strategies for B-cell aging can follow the principles of “deplete age-promoting subsets, preserve beneficial populations, and rebuild function.” First, selectively remove age-associated B cells (Camell et al., 2019) (ABCs)—which are tightly linked to immunosenescence and inflammaging—by blocking the TLR7/9–IL-21/IFN-γ axis required for their differentiation or by employing targeted depletion approaches, while sparing the normal B-cell compartment. Second, target aged adipose B cells (AABs), which express high levels of IL-1R and are induced by IL-1β/IL-18: anti-CD20 antibodies or an IL-1 receptor antagonist (IL-1RA) can reduce AAB abundance and improve lipolysis and metabolic profiles in mice. Third, maintain and restore the antimicrobial “housekeeping” function of B-1 cells and their IgM secretion to counteract age-related contraction of the IgM repertoire and affinity, thereby strengthening primary responses to novel pathogens and vaccines. When necessary, a short-term, global B-cell “reset” using a CD19/B220/CD22 depletion cocktail may reactivate bone-marrow B lymphopoiesis and rejuvenate the peripheral pool; however, this strategy alone has not restored immune competence or vaccine responsiveness and should be combined with the above selective depletion and functional reconstruction measures. Integration with precision vaccination and individualized monitoring is recommended to enhance protection while managing infection risk and potential immunosuppression. Larger, well-controlled studies are needed to establish long-term safety and efficacy (Keren et al., 2011).
3.3 Macrophage
Framed around the triad of “restoring sensing—rebalancing polarization—promoting resolution,” the strategy can be summarized as follows (Jackaman et al., 2013): (i) Immune reprogramming: In aged mice, targeted IL-2/anti-CD40 combination therapy can re-enable macrophage capacity to activate T cells and to initiate M1-like inflammatory programs, indicating that functional reinstatement via co-stimulatory/cytokine axes is feasible. (ii) Metabolic and autophagy targeting: Aged macrophages exhibit reduced autophagy, dysregulated nutrient sensing, and mitochondrial dysfunction. Caloric restriction and its mimetics (Fabbiano et al., 2016; Fahy et al., 2019) (e.g., metformin, resveratrol, rapamycin) may improve energy metabolism and autophagy, modulate mTOR/AMPK pathways, correct M1/M2 imbalance, and dampen chronic inflammation, thereby potentially extending healthspan and enhancing innate immune competence. (iii) Pro-resolution enhancement: Delayed inflammatory resolution in older adults is linked to hyperactive p38 signaling and aberrant TIM-4 expression (De Maeyer et al., 2020). Oral p38 inhibition can normalize TIM-4-mediated efferocytosis and the resolution program and, by curbing CCL2-dependent recruitment of inflammatory monocytes, reduce local inflammation while augmenting antigen-specific responses. This approach is a promising option for age-related inflammatory diseases, though larger studies are required to establish efficacy and safety. (iv) Foundational functional support: In conjunction with the above, measures to counter diminished TLR expression and insufficient MHC-II induction should be implemented to bolster pathogen recognition and antigen presentation as the bedrock of a comprehensive regimen.
Overall, we recommend a core framework of “metabolic correction - precise pathway modulation - resolution promotion,” applied in individualized combinations, with ongoing monitoring for infection risk, potential immunosuppression, and metabolic adverse effects.
3.4 Other
Immunogeroprotective strategies can be grouped into four categories. (i) (Sun et al., 2010) Thymic regeneration: Centered on FOXN1, approaches include gene therapy (Oh et al., 2020), transplantation of FOXN1-high thymic epithelial stem cells (Campinoti et al., 2020), somatic cell reprogramming to generate ectopic thymus, and decellularized scaffolds repopulated with human cells to restore thymic architecture and naïve T-cell output; adjunctive factors such as KGF, IGF-1, BMP4, and IL-7/IL-22 may further enhance function (Chu et al., 2008; Min et al., 2007; Wertheimer et al., 2018). Because benefits may be offset by fibrosis of secondary lymphoid organs, antifibrotic or senolytic agents—or synthetic lymph nodes to remodel the microenvironment—should be considered in combination. (ii) Targeting immune metabolism: Caloric restriction (CR) and its mimetics attenuate inflammaging by inhibiting mTOR, activating AMPK/autophagy and sirtuins, and improving T and NK-cell function. Representative agents include metformin (which improves mitochondrial function and autophagy in aged CD4^+^ T cells; broad epidemiologic benefits; TAME in progress) (Cabreiro et al., 2013; Partridge et al., 2020), rapamycin (Strong et al., 2016) and other mTORC1 inhibitors (enhanced influenza vaccine responses, though phase III results have been mixed), resveratrol, spermidine, and α-ketoglutarate; infection risk and population-specific evidence must be balanced. (iii) Senescent-cell clearance (Xu et al., 2018): Senolytics that target anti-apoptotic pathways or β-galactosidase-activated prodrugs reduce the SASP and systemic inflammation (Amor et al., 2020); CAR-T cells engineered against markers such as uPAR offer targeted elimination. (iv) Epigenetic modulation: Interventions addressing aberrant DNA methylation (Garg et al., 2014) and chromatin states, suppressing retrotransposon activation (De Cecco et al., 2019; Simon et al., 2019) (e.g., via SIRT6 activation), and exploring DNMT/HDAC-directed agents may mitigate immunosenescence.
Overall, a combinatorial, stratified, and evidence-based program—integrating organ regeneration, metabolic reprogramming, senescent-cell clearance, and epigenetic modulation—is recommended, with vigilant monitoring for immune tolerance, tumor immunosurveillance trade-offs, and metabolic adverse effects, and with dedicated trials in healthy older adults versus disease cohorts plus long-term follow-up. The pathogenic mechanism and the corresponding treatment methods are shown in Table 1.
4 Conclusion and outlook
Coronary artery disease (CAD) is closely linked to immunosenescence, whereby age-related decline in immune function promotes chronic inflammation, vascular injury, and atherosclerosis. Through dysfunction of multiple immune cell populations—including T cells, B cells, and macrophages—immunosenescence attenuates immune responsiveness and sustains persistent inflammatory activity. In recent years, several intervention strategies targeting immunosenescence have shown preliminary promise, including restoration of hematopoietic stem-cell function, activation and rejuvenation of T- and B-cell compartments, modulation of macrophage activity, thymic regeneration, and immunometabolic reprogramming; together, these approaches offer new therapeutic directions for CAD. Future research should prioritize elucidating the mechanistic roles of immunosenescence in CAD, developing precise, personalized treatment strategies, and validating their safety and efficacy in large-scale clinical trials. Combination regimens that integrate immune modulation with metabolic interventions are likely to represent an important therapeutic avenue, with the potential to improve treatment efficacy and long-term patient outcomes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Allsopp R. C.Morin G. B.De Pinho R.Harley C. B.Weissman I. L. (2003). Telomerase is required to slow telomere shortening and extend replicative lifespan of HS Cs during serial transplantation. Blood 102 (2), 517–520. 10.1182/blood-2002-07-2334 12663456 · doi ↗ · pubmed ↗
- 2Álvarez-Álvarez M. M.Zanetti D.Carreras-Torres R.Moral P.Athanasiadis G. (2017). A survey of sub-saharan gene flow into the mediterranean at risk loci for coronary artery disease. Eur. J. Hum. Genet. 25 (4), 472–476. 10.1038/ejhg.2016.200 28098150 PMC 5386420 · doi ↗ · pubmed ↗
- 3Álvarez-Heredia P.Reina-Alfonso I.Domínguez-Del-Castillo J. J.Gutiérrez-González C.Hassouneh F.Batista-Duharte A. (2023). Accelerated T-Cell immunosenescence in cytomegalovirus-seropositive individuals after severe acute respiratory syndrome coronavirus 2 infection. J. Infect. Dis. 228 (5), 576–585. 10.1093/infdis/jiad 119 37103009 PMC 10469128 · doi ↗ · pubmed ↗
- 4Amor C.Feucht J.Leibold J.Ho Y. J.Zhu C.Alonso-Curbelo D. (2020). Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583 (7814), 127–132. 10.1038/s 41586-020-2403-9 32555459 PMC 7583560 · doi ↗ · pubmed ↗
- 5Basatemur G. L.Jørgensen H. F.Clarke M. C. H.Bennett M. R.Mallat Z. (2019). Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 16 (12), 727–744. 10.1038/s 41569-019-0227-9 31243391 · doi ↗ · pubmed ↗
- 6Benjamin E. J.Blaha M. J.Chiuve S. E.Cushman M.Das S. R.Deo R. (2017). Heart disease and stroke Statistics-2017 update: a report from the American heart association. Circulation 135 (10), e 146–e 603. 10.1161/cir.0000000000000485 28122885 PMC 5408160 · doi ↗ · pubmed ↗
- 7Bergström I.Backteman K.Lundberg A.Ernerudh J.Jonasson L. (2012). Persistent accumulation of interferon-γ-producing CD 8+CD 56+ T cells in blood from patients with coronary artery disease. Atherosclerosis 224 (2), 515–520. 10.1016/j.atherosclerosis.2012.07.033 22882906 · doi ↗ · pubmed ↗
- 8Binder C. J.Papac-Milicevic N.Witztum J. L. (2016). Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 16 (8), 485–497. 10.1038/nri.2016.63 27346802 PMC 7097710 · doi ↗ · pubmed ↗