Coexistence of Acute Demyelinating Polyneuropathy and LRP4-Positive Myasthenia Gravis
Halit Fidancı, Sevgi Turhan, Halil Can Alaydın, Ahmet Yusuf Ertürk, İlker Öztürk

TL;DR
This paper reports a rare case where a patient had two autoimmune disorders, GBS and LRP4-positive MG, highlighting the diagnostic challenges and treatment approaches.
Contribution
The paper presents a rare case of coexisting acute demyelinating polyneuropathy and LRP4-positive myasthenia gravis, emphasizing the need for comprehensive diagnostic evaluation.
Findings
A patient with acute demyelinating polyneuropathy later developed LRP4-positive myasthenia gravis.
Comprehensive electrophysiological and antibody testing is crucial for diagnosing overlapping autoimmune conditions.
Treatment with plasmapheresis and immunosuppressants improved symptoms of both disorders.
Abstract
Guillain–Barré syndrome (GBS) and myasthenia gravis (MG) are rare autoimmune disorders that may share overlapping features such as ophthalmoparesis, limb weakness, and bulbar symptoms, complicating the differential diagnosis. Coexistence of GBS and MG or chronic inflammatory demyelinating polyneuropathy and MG, particularly the low-density lipoprotein receptor-related protein 4 (LRP4) antibody-positive subtypes, is extremely rare. We present such a case to highlight diagnostic challenges. A 46-year-old man presented with distal weakness, sensory loss, facial diplegia, and dyspnea. Nerve conduction studies revealed demyelinating features, and cerebrospinal fluid analysis showed albuminocytologic dissociation. An acute demyelinating polyneuropathy, most likely GBS, was suspected, and clinical improvement was observed following plasmapheresis. Three weeks later, new symptoms including…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
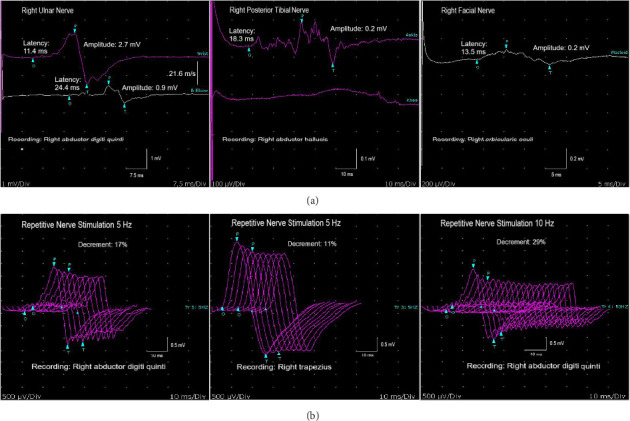 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMyasthenia Gravis and Thymoma · Peripheral Neuropathies and Disorders · Parkinson's Disease and Spinal Disorders
1. Introduction
Guillain-Barré syndrome (GBS) and myasthenia gravis (MG) are rare autoimmune disorders involving the peripheral nervous system and the neuromuscular junction, with incidence rates of 1-2 and 1-3 per 100,000, respectively [1–3]. GBS may present with limb weakness, reduced deep tendon reflexes, and bulbar symptoms such as facial weakness and dysphagia. MG is characterized by muscle weakness, often presenting as fluctuating ptosis or ophthalmoparesis. Differentiation between GBS and MG relies on clinical presentation, electrodiagnostic studies, and laboratory findings [1–3]. Since ophthalmoparesis, limb weakness, and bulbar symptoms can be seen in both MG and GBS, their coexistence may complicate the differential diagnosis. Furthermore, the occurrence of ptosis, usually regarded as typical for MG, in GBS subgroups such as Miller Fisher syndrome, may blur the clinical distinction between these conditions [1–3].
Although these two autoimmune neuromuscular disorders affect different regions, the coexistence of GBS and MG (GBS-MG) has been rarely reported [4–8]. Similar to the reported association between GBS and MG, a relationship between chronic inflammatory demyelinating polyneuropathy (CIDP) and MG has also been documented [9]. To highlight this unusual association, we present a patient initially diagnosed with acute demyelinating polyneuropathy who was subsequently found to have MG, specifically the low-density lipoprotein receptor-related protein 4 (LRP4) antibody-positive subtype.
2. Case Presentation
A 46-year-old male patient described sensory symptoms and weakness in the distal lower extremities starting about 1 week before his presentation. He stated that tingling sensory symptoms and weakness developed in the distal upper extremities in addition to the distal lower extremities 2-3 days after the onset of symptoms. There was no history of recent infection, trauma, vaccination, or diarrhea before the onset of symptoms. His medical history included a 15 year history of diabetes mellitus, coronary artery disease, and chronic alcohol consumption. He denied any prior history of ptosis, diplopia, or dysphagia. In his neurological examination, he had significant hypoesthesia in the distal extremities, mild bilateral facial weakness, and dyspnea. Muscle strength was symmetrically graded as 3 in both distal and proximal muscles of the lower extremities and as 4 in the distal muscles of the upper extremities with minimal proximal weakness, according to the Medical Research Council (MRC) scale. Neurological assessment revealed absent deep tendon reflexes. The patient stated that his symptoms showed no fluctuation during the day, which was consistent with the neurological examination findings. He reported mild dyspnea and was able to reach 20 on the single-breath count test. Bladder and bowel functions were preserved. In sensory nerve conduction studies (NCSs), bilateral median, ulnar and sural compound nerve action potentials (CNAPs) could not be obtained. Motor NCSs of the right median, ulnar, and bilateral posterior tibial nerves showed features of demyelination, including prolonged latencies, slowed velocities, and conduction blocks (Table 1). Positive sharp waves and fibrillation potentials, along with a reduced recruitment pattern, were observed in the right first dorsal interosseous and tibialis anterior muscles. Myopathic motor unit action potentials were not present. The protein level in the cerebrospinal fluid was 1198 mg/L with lumbar puncture and no cells were found. GQ1b and GM1 antibodies were found negative. Serum electrolytes, vitamin b12, folate, and vitamin D levels, as well as liver, renal, and thyroid function tests, were within normal limits. The patient underwent plasmapheresis five times. Muscle strength improved to grade 4 in the distal muscles of the lower extremities with minimal proximal weakness, and grade 5 in the upper extremities upon discharge. There was a significant improvement in the patient's facial weakness, and he reported no further dyspnea.
Approximately 3 weeks later, the patient presented with dysarthria, hypernasal speech, and difficulty swallowing. Compared to the initial visit, the patient exhibited more marked dyspnea and bilateral facial weakness. On reassessment, the patient was able to count only 9-10 on a single-breath. As in the initial study, CNAPs of the median, ulnar, and sural nerves were absent. Motor NCS results are presented in Table 1, and Figure 1(a) illustrates the right ulnar, posterior tibial, and facial motor NCS findings. A decremental response of more than 10% was found in 2, 3, 5, 10 Hz repetitive nerve stimulation (RNS) recorded from the right abductor digiti quinti (ADM) and trapezius muscles (Figure 1(b)). No significant facilitation was observed with exercise in the CMAP recorded from the right abductor pollicis and ADM muscles. Jitter abnormality could not be evaluated, as the patient was unable to tolerate the procedure. Antibodies against the acetylcholine receptor (AchR) and muscle-specific tyrosine kinase were negative, whereas the LRP4 antibody was positive. Thoracic computed tomography revealed no evidence of thymoma. Lumbar puncture was not carried out during the second presentation. The patient was given intravenous immunoglobulin for 5 days. Treatment was initiated with pyridostigmine, oral steroids, and azathioprine. At the examination conducted 3 weeks after discharge, the patient's dysarthria and speech and swallowing difficulties resolved; muscle strength was graded as 4 in the distal lower extremities, while strength in all other muscle groups was normal.
3. Discussion
GBS-MG is extremely rare, with an estimated incidence of approximately one in a billion, and has been discussed in only a very limited number of case reports [4–7]. However, this estimate may be low when acute motor axonal neuropathy (AMAN) is considered, as the diagnosis can be missed in some patients. As illustrated in this case, some patients initially considered to have relapses or treatment-related fluctuations may, in fact, have GBS-MG or coexistence of acute-onset CIDP and MG.
The pathophysiological mechanism underlying GBS-MG has not been clearly elucidated; however, it may involve antigens or antibodies, triggered by infection, thymic abnormalities, or other factors, that target both postsynaptic membrane proteins such as AchR and peripheral nervous system components such as sphingomyelin [4, 6, 7]. In light of the current case with LRP4 positivity, one possible explanation is that a trigger or antigen induces antibodies that, via molecular mimicry, cross-react with both peripheral nerves and LRP4. In addition, uncovering the relationship between LRP4 and peripheral nerve regeneration may help explain the association between LRP4-positive MG and GBS [8].
AchR antibodies are commonly identified in patients with MG; in contrast, LRP4 antibody positivity is observed in approximately 1%–3% of all cases and may be associated with a milder clinical course [2, 3]. This may explain the absence of MG-related symptoms in the present case prior to presentation. While GBS-MG has been documented, the absence of AchR antibodies in this case may be attributed to prior plasmapheresis. Although this poses a diagnostic limitation, it is important to note that the antibody testing was performed approximately 3 weeks after the last plasmapheresis session. AchR antibody levels may not have fully returned to baseline within this timeframe, but they are known to begin increasing by the second week postprocedure [10]. Furthermore, although diabetes mellitus and chronic alcohol use could confound the interpretation of NCSs in the diagnosis of GBS, the demyelinating characteristics identified in the NCSs support the presence of an acquired demyelinating polyneuropathy. The findings were suggestive of acute polyneuropathy, as the patient had no prior extremity weakness and reported an acute onset beginning in the lower limbs with subsequent spread. In addition, the prolonged onset latencies and reduced amplitudes observed in the second motor NCS compared to the first support the presence of an acute polyneuropathy. In the setting of chronic polyneuropathy, CMAP amplitudes would be unlikely to decrease to this extent within 4 weeks. While acute-onset CIDP remains a differential consideration, the presence of bilateral facial weakness and dyspnea makes this diagnosis less probable, though not entirely excluded [11]. Therefore, the coexistence of CIDP and MG is also a possibility [9]. At the patient's second admission, lumbar puncture was not performed; however, a decrease in cerebrospinal fluid protein could have suggested GBS.
4. Conclusion
We suggest that in patients diagnosed with GBS or acute demyelinating polyneuropathy, when clinical symptoms worsen again following initial improvement after plasmapheresis or IVIG, the possibility of GBS-MG or coexisting acute-onset CIDP and MG should be considered in the differential diagnosis, alongside treatment-related fluctuations or relapses. We also recommend that RNS be performed and that additional antibodies, such as LRP4, are tested along with AchR antibodies, especially in the context of suspected GBS-MG or coexistence of acute-onset CIDP and MG.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bellanti R. Rinaldi S. Guillain-Barré Syndrome: A Comprehensive Review European Journal of Neurology 2024318 p. e 1636510.1111/ene.16365 PMC 1123594438813755 · doi ↗ · pubmed ↗
- 2Gilhus N. E. Myasthenia Gravis New England Journal of Medicine 2016375262570258110.1056/NEJ Mra 16026782-s 2.0-8500729320428029925 · doi ↗ · pubmed ↗
- 3Hehir M. K. Silvestri N. J. Generalized Myasthenia Gravis: Classification, Clinical Presentation, Natural History, and Epidemiology Neurologic Clinics 201836225326010.1016/j.ncl.2018.01.0022-s 2.0-8504556081729655448 · doi ↗ · pubmed ↗
- 4Kraus J. Teismann I. Kellinghaus C. Temporal Coincidence Between AMAN Type of GBS and Myasthenia Gravis Journal of Neurology 2007254226426510.1007/s 00415-006-0366-x 2-s 2.0-3384763682717308867 · doi ↗ · pubmed ↗
- 5Karri M. Ramasamy B. Perumal S. Occurrence of Guillain-Barré Syndrome and Myasthenia Gravis in an Elderly Male The Egyptian Journal of Neurology, Psychiatry and Neurosurgery 2019551 p. 8310.1186/s 41983-019-0136-1 · doi ↗
- 6Yuan J. Zhang J. Zhang B. Hu W. The Clinical Features of Patients Concurrent With Guillain-Barré Syndrome and Myasthenia Gravis Neurosciences 2018231667010.17712/nsj.2018.1.201702092-s 2.0-8504106074829455227 PMC 6751915 · doi ↗ · pubmed ↗
- 7Cao Y. Gui M. Ji S. Bu B. Guillain-Barré Syndrome Associated With Myasthenia Gravis: Three Cases Report and a Literature Review Medicine (Baltimore) 20199847 p. e 1810410.1097/MD.0000000000018104 PMC 688260831764848 · doi ↗ · pubmed ↗
- 8Hui T. K. Lai X. S. Dong X. Ablation of Lrp 4 in Schwann Cells Promotes Peripheral Nerve Regeneration in Mice Biology 2021106 p. 45210.3390/biology 10060452 PMC 822397634063992 · doi ↗ · pubmed ↗