Acquired MTAP Loss Following Entrectinib Resistance in ROS1‐Rearranged NSCLC With CD74 Exon 3–ROS1 Exon 34 Fusion
Mizuha Haraguchi Hashiguchi, Maika Tanino, Suzuyuki Yoneda, Masato Asaoka, Junko Kagyo, Makoto Katayama, Hideki Terai, Kohei Nakamura, Hiroshi Nishihara, Koichi Fukunaga

TL;DR
A patient with ROS1-rearranged lung cancer developed resistance to entrectinib, linked to acquired MTAP loss despite no known resistance mutations.
Contribution
Identifies MTAP loss as a novel resistance mechanism in ROS1-rearranged NSCLC following entrectinib treatment.
Findings
Acquired MTAP loss was observed in a ROS1-rearranged NSCLC patient after entrectinib resistance.
The CD74–ROS1 fusion remained unchanged, with no known resistance mutations detected.
Longitudinal genomic profiling is critical for understanding resistance mechanisms and treatment decisions.
Abstract
This case highlights acquired MTAP loss during disease progression in ROS1‐rearranged NSCLC. Despite persistent CD74–ROS1 fusion and absence of known resistance mutations, the patient developed CNS progression after entrectinib, underscoring the value of longitudinal genomic profiling in guiding treatment decisions.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
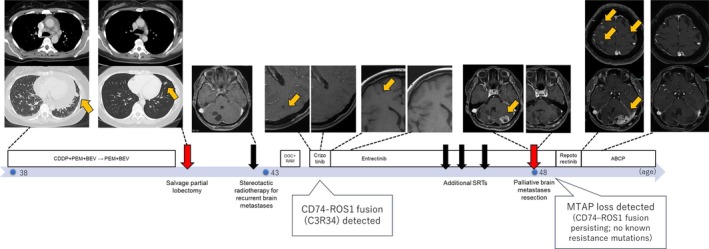 Figure 1
Figure 1 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer-related gene regulation · Epigenetics and DNA Methylation · Histone Deacetylase Inhibitors Research
We previously reported a lung adenocarcinoma case in a 38‐year‐old never‐smoking female harboring a novel CD74 exon 3–ROS1 exon 34 (C3R34) fusion, demonstrating a marked response to entrectinib [1]. Here, we present the clinical course focusing on the emergence of acquired MTAP (methylthioadenosine phosphorylase) loss at age 48 despite persistent ROS1 fusion positivity.
At 43 years, after postoperative recurrence, comprehensive genomic profiling (FoundationOne) confirmed the CD74–ROS1 (C3R34) fusion [1]. Entrectinib treatment achieved durable disease control for approximately 4 years. During this period, a new cerebellar lesion appeared and was initially managed with stereotactic radiotherapy, but eventually progressed, causing cerebellar symptoms. Surgical resection at 48 years confirmed viable adenocarcinoma.
In‐house hybridization capture‐based CGP (PleSSision Rapid) [2] revealed ongoing CD74–ROS1 fusion. Importantly, no known secondary ROS1 resistance mutations (such as G2032R, D2033N, S1986F/Y, or L2026M), nor bypass pathway alterations (including EGFR, KRAS, BRAF, MET, and PIK3CA), were detected. However, newly acquired MTAP loss was identified. Given the clinical progression and available genomic data, therapy was switched to repotrectinib; however, the patient showed no clinical benefit and developed neurological deterioration with diplopia and abducens nerve palsy. MRI showed local recurrence, multifocal brain metastases, and leptomeningeal enhancement. Subsequent radiotherapy was given for symptom palliation, followed by ABCP chemotherapy (atezolizumab, bevacizumab, carboplatin, and paclitaxel). After four cycles, imaging demonstrated stabilization of irradiated lesions and regression of non‐irradiated metastases. A timeline of the clinical course is shown in Figure 1.
MTAP loss has been implicated in tumor aggressiveness and resistance mechanisms across cancers. This case raises the possibility that MTAP loss may be associated with disease progression under ROS1‐targeted therapy, although causality cannot be established. Importantly, MTAP deficiency may confer sensitivity to pemetrexed [3] and PRMT5 inhibitors [4], indicating possible therapeutic avenues in refractory ROS1‐positive NSCLC. This report is limited by its single‐case nature, which precludes generalizability. Moreover, while MTAP loss was detected at progression, this temporal association does not prove causality, and unrecognized resistance mechanisms may have contributed. Nevertheless, this case highlights that longitudinal molecular profiling of recurrent lesions, including CNS sites, may provide actionable insights for treatment selection in advanced NSCLC.
Author Contributions
Mizuha Haraguchi Hashiguchi: conceptualization, writing – original draft, writing – review and editing. All other authors: writing – review and editing.
Ethics Statement
The authors have nothing to report.
Consent
Written informed consent was obtained from the patient for publication of all clinical and genomic information presented in this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1M. H. Hashiguchi , T. Sato , R. Watanabe , et al., “A Case of Lung Adenocarcinoma With a Novel CD 74–ROS 1 Fusion Variant Identified by Comprehensive Genomic Profiling That Responded to Crizotinib and Entrectinib,” Thoracic Cancer 12, no. 18 (2021): 2504–2507, 10.1111/1759-7714.14093.34319660 PMC 8447907 · doi ↗ · pubmed ↗
- 2H. Takaoka , H. Terai , K. Nakamura , et al., “Clinical Application of In‐House Comprehensive Genomic Profiling for Thoracic Cancer: Insights From a Japanese Hospital,” Cancer Science 116 (2025): 2819–2830, 10.1111/cas.70168.40757605 PMC 12485672 · doi ↗ · pubmed ↗
- 3O. Alhalabi , J. Chen , Y. Zhang , et al., “MTAP Deficiency Creates an Exploitable Target for Antifolate Therapy in 9p 21‐Loss Cancers,” Nature Communications 13, no. 1 (2022): 1797, 10.1038/s 41467-022-29397-z.PMC 898001535379845 · doi ↗ · pubmed ↗
- 4G. V. Kryukov , F. H. Wilson , J. R. Ruth , et al., “MTAP Deletion Confers Enhanced Dependency on the PRMT 5 Arginine Methyltransferase in Cancer Cells,” Science 351, no. 6278 (2016): 1214–1218, 10.1126/science.aad 5214.26912360 PMC 4997612 · doi ↗ · pubmed ↗