Translational regulation of human papillomavirus mRNAs in carcinogenesis: old questions and new insights
Noemi Baranda-Ávila, Giovanna Maldonado, Dora E. Vélez, Greco Hernández

TL;DR
This paper reviews how human papillomavirus mRNAs are translated during cancer development, highlighting gaps in understanding and potential treatment strategies.
Contribution
The paper introduces new hypotheses about IRES-mediated translation and emphasizes the role of translational regulation in HPV-related carcinogenesis.
Findings
Translation mechanisms for E5, L1, and L2 proteins remain unclear.
Viral-cellular chimeric transcripts' translation is not well understood.
Translational regulation is critical during epithelial cell differentiation in HPV-induced cancer.
Abstract
Persistent infection with high-risk human papillomaviruses (HR-HPVs) is a major etiological factor in the development of cervical cancer, which ranks as the fourth leading cause of cancer-related mortality among women worldwide. HR HPV types 16 and 18 cause more than 70% of all cases. These viruses encode the proteins E1, E2, E1^E4, E4, E5, E6, E7, L1, and L2, through a complex array of polycistronic mRNAs. For decades, research on HPV gene expression has focused predominantly on transcriptional activity and mRNA splicing. In contrast, the mechanisms underlying the translation of mRNAs remain poorly understood. Whereas the translational regulation of E1, E2, E6, and E7 has been elucidated, the translation mechanisms for E5, L1, and, L2 proteins are still unclear. We hypothesized that their translation may occur via internal ribosome entry sites (IRESs). Other critical questions also…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
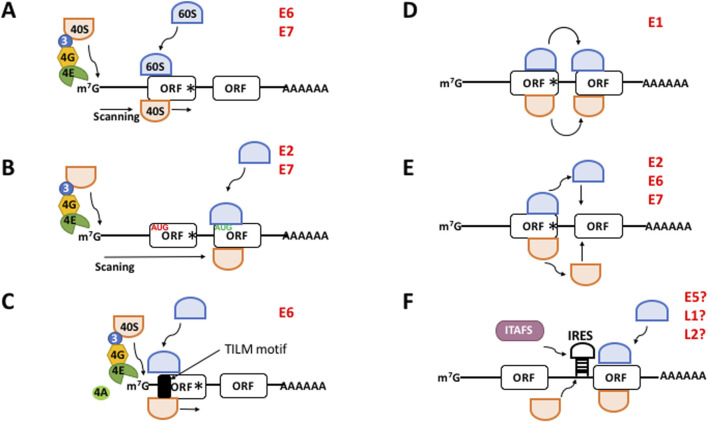 FIGURE 1
FIGURE 1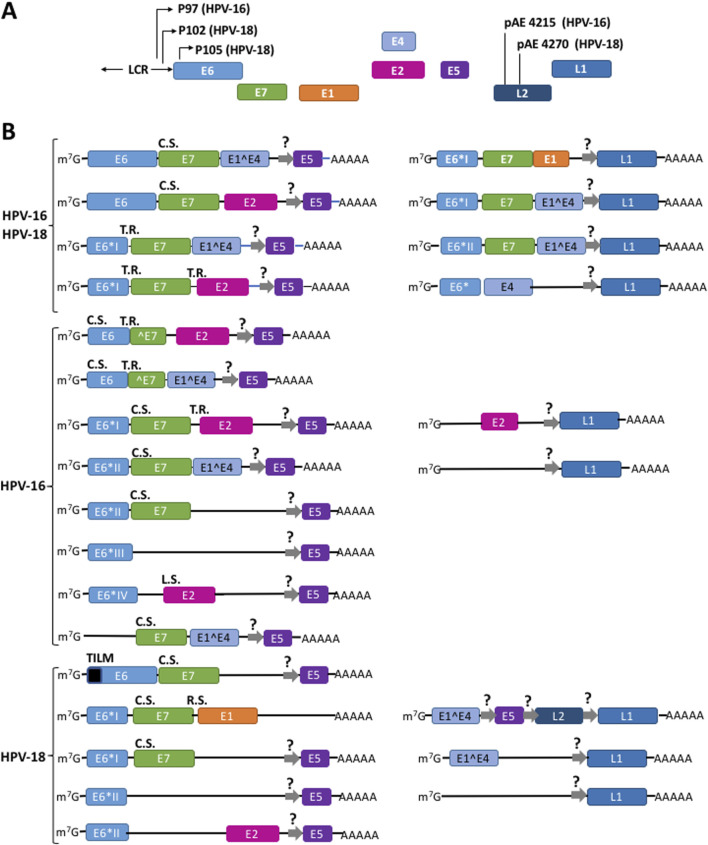 FIGURE 2
FIGURE 2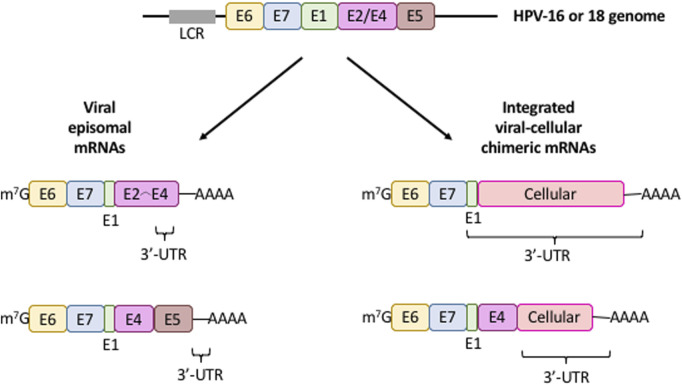 FIGURE 3
FIGURE 3| Virus | Cistron | Mechanism | Reference |
|---|---|---|---|
| VPH-16 | E1 | Not known | |
| E1^E4 | Not known; possible IRES | This review | |
| E2 | Termination-reinitiation or leaky scanning |
| |
| E5 | Not known; possible IRES |
| |
| E6 | Canonical |
| |
| E7 | Termination-reinitiation or leaky scanning |
| |
| L1 | Not known; possible IRES | This review | |
| L2 | Not known | ||
| VPH-18 | E1 | Ribosome shunting |
|
| E1^E4 | Not known; possible IRES | This review | |
| E2 | Not known | ||
| E5 | Not known; possible IRES |
| |
| E6 | Scanning-independent using extremely short (0–3 nucleotides long) 5′-UTRs and an RNA sequence within the ORF; it also requires the mRNA cap and poly(A) tail; it requires eIF4E and eIF4A |
| |
| E7 | Termination-reinitiation or canonical |
| |
| L1 | Not known; possible IRES | This review | |
| L2 | Not known; possible IRES | This review |
| Virus | Cellular gene fussed with the viral E6,E7 mRNAs | Biological consequence on cancer | Sample source | Integration site | Reference |
|---|---|---|---|---|---|
| HPV-16 | Non-determined | Integration of HPV-16 DNA and expression of virus-cell mRNAs results in the increased stability of E6 and E7 mRNAs and cellular growth | W12 clonal populations and cervical tumor cell lines (SiHa, CaSki) | HPV integration does not occur at any specific locus on the host genome |
|
| HPV-16 |
| Viral-cellular fusion transcripts are more stable than episome-derived transcripts, promoting carcinogenesis | Cervical carcinoma tumors |
|
|
| HPV-16 |
| HPV integration is a random process that promotes selection of aggressively expanding cells. Some of those i9ntergation events may hit oncogenes with their concomitant deregulation | HNC tumors, human keratinocytes derived from primary tonsillar or foreskin epithelia immortalized |
|
|
| HPV-16, HPV-18 | Non-determined | HPV-16/-18 DNA integration into host cell genome is related to the progression stage of vulvar dysplasia | VIN lesions and cervical carcinoma cell lines (SiHa and Caski) | Non-determined |
|
| HPV-16, HPV-18, HPV-31, HPV-33, HPV-35 |
| There is no correlation between HPV type and specific integration loci. HPV integration may occur in transcriptionally active regions and can influence on host gene expression | Ca, CIN3 |
|
|
| HPV-2, HPV-6, HPV-11, HPV-13, HPV-16, HPV-18, HPV-30, HPV-31, HPV-35 | Non-determined | The 3′ region HPV-16 mRNA represses the expression at both mRNA and protein level. For other HPV types, except 18, the 3′ region represses mRNA expression by posttranscriptional mechanisms | HaCaT and SiHa cells | Non-determined |
|
| HPV-16, HPV-18 | N- | Integration of HPV genome results in overexpression of the oncogene c- | Invasive genital carcinomas | Chromosome band 8q24.1, nearby the protooncogene c- |
|
| HPV-16, HPV-18 | Non-determined | The steady-state levels of c- | Primary Ca and Ca cells (SiHa, SW756, HeLa, and C4-1) | Chromosome regions 20pter->20ql3 and 3p25->3qter, nearby the protooncogenes c- |
|
| HPV-16 | Non-determined | Non-determined | Ca and normal placentas | No cellular sequence specific for the integration is found; some samples showed |
|
| HPV-16 | Non-determined | Non-determined | Keratinocyte SK-v cells; intraepithelial neoplasms termed bowenoid papulas | Non-determined |
|
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA modifications and cancer · RNA Research and Splicing · Cancer-related molecular mechanisms research
Introduction
Cervical cancer ranks fourth in incidence and third in mortality among neoplasias affecting women worldwide. Its burden might reach highest mortality rates in more than 40 low-income countries (National Cancer Institute, 2021; LaVigne et al., 2017; Sung et al., 2021; WHO, 2022). HPV prevalence is markedly higher in emerging regions, reaching 42.2% compared to 22.6% in high-income regions (Pavelescu et al., 2025). Persistent infection of the genital tract with high-risk (HR) human papillomavirus (HPV) is the primary cause of most cervical cancer cases. Among these, HR-HPV types 16 and 18 are responsible for over 70% of cases. Moreover, about 90% of anal cancer, 75% of vaginal cancer, 70% of oropharyngeal and vulvar cancers, and 63% of penile cancers are caused by HR-HPVs across the globe (Alteri et al., 2025). Effective vaccines against HR-HPV have been developed, and HPV vaccination was introduced in 73 countries during 2010–2019 and in 30 additional countries during 2020–2023. Global first-dose HPV coverage among girls increased from 17% in 2019 to 27% in 2023, and full-course coverage rose from 13% in 2019 to 20% in 2023 (Jones et al., 2024). According to World Health Organization, global first-dose coverage further increased to 31% in 2024. However, this is still far below the 2030 target of 90% coverage because their use remains limited or non-existent in many countries (Collaborators, 2025), highlighting cervical cancer as a major public health challenge worldwide.
HR-HPVs infect basal keratinocytes of the mucosal epithelium, leading to the formation of low-grade intraepithelial lesions. Once within the host cells, the viral genome replicates as an extrachromosomal DNA inside the cell nucleus. As epithelial cells divide and differentiate, HPV gene expression couples with cell differentiation, and cells progress to productive viral states, leading to cell proliferation, immortalization, and the development of malignant phenotypes (Nelson and Mirabello, 2023; Obanya et al., 2025; Pavelescu et al., 2025; Rosendo-Chalma et al., 2024). This process has been extensively studied with regard to the transcription and splicing of viral mRNAs (Doomer et al., 2024; Olmedo-Nieva et al., 2018; Wang et al., 2025; Yu et al., 2022) (https://pave.niaid.nih.gov/#explore/transcript_maps). However, the translation of viral mRNAs remains the least studied aspect of viral gene expression (Kajitani and Schwartz, 2022).
Notably, translational control is emerging as a key process of viral proteins synthesis, opening novel avenues to target the translational machinery to combat HVP-induced cervical cancer. We review the old, yet unresolved questions regarding the translation of viral mRNAs, alongside recent advances in our understanding of viral protein synthesis regulation and their implications for the design of new therapeutic strategies.
Translation in eukaryotes
Translation, i.e., the decoding of genetic information of a mRNA into peptide by the ribosome and translation factors using a genetic code, largely determines in all cells protein abundance and the proteome composition in normal physiology, stresses and diseases (Hernández and Tettweiler, 2012). Accordingly, a myriad of mechanisms to regulate mRNA translation exists in cells and virus (Hernández et al., 2020; Hershey et al., 2019; Jan et al., 2016; Kwan and Thompson, 2019). Translation consists of four main stages, namely initiation, elongation, and termination. In some physiological situations a fourth stage may happen for some mRNAs, i.e., ribosome recycling. However, the global process of translation is largely controlled at the initiation step (Hershey et al., 2019). Different mechanism account for the initiation step, namely the canonical mechanism and the non-canonical leaky scanning, ribosome shunting, termination-reinitiation, and IRES-dependent mechanisms.
The canonical mechanism to initiate translation
Most eukaryotic mRNAs initiate translation in the canonic manner, termed cap-dependent mechanism. It begins with the mRNA recruitment via recognition of the cap structure (7-methyl guanosine) at the 5′ of the mRNA by eukaryotic initiation factor (eIF) 4E. During this process, eIF4G binds eIF4E, eIF4A and poly(A)-binding protein (PABP). Subsequently, a free 40S ribosomal subunit in complex with eIF1, eIF1A, eIF3, eIF5 and a ternary complex (TC, consisting of eIF2 bound to an initiator Met-tRNA_i_ ^Met^ and GTP) forms a 43S pre-initiation complex (PIC). eIF4G further interacts with the ribosome-bound eIF3 to promote recruitment of the 43S PIC to the mRNA 5′-untranslated region (5′-UTR) (Brito Querido et al., 2024; Pelletier and Sonenberg, 2019). After the mRNA is recruited, the 43S PIC scans the 5′-UTR to reach the translation initiation site (TIS), most frequently an AUG codon (Figure 1A) (Brito Querido et al., 2024; Kwan and Thompson, 2019; Pelletier and Sonenberg, 2019). A context sequence surrounding the TIS, termed the “Kozak motif”, is required for optimal AUG codon recognition by the ribosomal complex. For human mRNAs, this motif consists of the sequence GCCA/GCCAUGG (Hernández et al., 2019). AUG recognition triggers 60S ribosomal subunit joining to assemble an 80S initiation complex which enters in successive rounds of peptide elongation (Brito Querido et al., 2024; Kwan and Thompson, 2019; Pelletier and Sonenberg, 2019). When a stop codon is recognized in the A site of the ribosome eukaryotic releasing factor (eRF) 1 promotes peptide liberation.
*The different mechanisms to initiate translation in eukaryotes. (A) Canonical, cap-, eIF4E-, and scanning dependent mechanism. (B–F) Non-canonical mechanisms. (B) Leaky scanning. mRNAs containing a weak Kozak AUG contexts (in red) allow the ribosomal 40S subunit to skip the first ORF and recognize the downstream AUG (in green) of the next ORF which is in good context. (C) Translation Initiation of Leaderless mRNAs (TILM)-dependent mechanism. Extremely short 5′-UTR and a TILM motif within the E6 ORF promote translation initiation. (D) Ribosome shunting. The 40S ribosome shunts over a transcript segment landing downstream to reach the AUG codon of the next ORF. (E) Termination-reinitiation. After translation has terminated ribosomes quit the mRNA. Then a new ternary complex is loaded onto the 40S ribosome and it resumes scanning to start translation of the next ORF. (F) IRES-dependent mechanism. The 40S ribosome directly binds an IRES next to the AUG start codon to initiate translation. This process may need the help of IRES-trans acting factors (ITAFS). m
7
G, methyl-7 GTP cap structure of mRNA; 40S, ribosomal subunit 40S; 60S, ribosomal subunit 60S; 3, eIF3; 4G, eIF4G; 4E, eIF4E; Black box, translation initiation of leaderless mRNAs (TILM) motif; ORF, open reading frame; AAAA, poly(A) tail; IRES, internal ribosome entry site. The viral proteins with a proposed mechanism are indicated in red. Hypothetical IRES-driven translation for E5, L1, and L2 is indicated with a?*
Non-canonical translation initiation mechanisms
In addition to the canonical pathway, translation initiation may happen via other mechanisms, which mostly account for the translation of internal ORF within polycistronic mRNAs in many viruses.
In the “leaky scanning” (Hinnebusch et al., 2004), mRNAs containing an unfavorable Kozak (i.e., “weak”) motif allow the ribosomal 43S PIC skip the first open reading frame (ORF) and recognize a downstream ORF AUG (Figure 1B). Recently, we demonstrated that E6,E7 mRNAs from HPV-18 possesses extremely short 5′-UTRs (0–5 nucleotides) in cervical tumors which drive the translation of E6 (García et al., 2021), conforming a novel non-canonical mechanism of translation initiation (Figure 1C). In this virus, E6 is translated in a scanning-independent manner but is dependent on the mRNA cap and the poly(A) tail, eIF4E, eIF4A, and on a novel RNA motif within the coding region of the E6 cistron that we termed TILM (for Translation Initiation of Leaderless mRNAs). Thus, in the absence of a 5′-UTR, TILM controls the translation of E6 (García et al., 2021). In the “ribosome shunting” (Kwan and Thompson, 2019), specific cis-elements within the mRNA promote that the 43S PIC bypasses or shunts over a segment of the transcript, lands downstream and resumes scanning to reach the TIS (Figure 1D). In the “termination-reinitiation” (Jackson et al., 2012), after translation has terminated, a new TC is loaded onto a 40S subunit and it resumes scanning to start the translation of a downstream ORF on the same mRNA (Figure 1E). For some other transcripts, translation initiation is driven by an internal ribosome entry site (IRES) (Kwan and Thompson, 2019), a cis-located mRNA region that promotes ribosomal recruitment just nearby the TIS in a cap- and eIF4E-independent manner and without scanning. This process may be promoted by the activity of cellular IRES-trans-acting factors (ITAFs) (Figure 1F).
Function of the viral proteins during infection and carcinogenesis
HPVs encode the early proteins E1, E2, E4, E5, E6, and E7 from the early (E) control region of the viral genome, and the capsid proteins L1 and L2 from the long control region (LCR) (Doorbar et al., 2015; Nelson and Mirabello, 2023; Rosendo-Chalma et al., 2024). E1 is a helicase that, in cooperation with E2, drives viral genome replication. E2 also regulates the viral cycle life and controls the transcription of the E6 and E7 oncogenes. E4 plays a role in virion assembly and facilitates the release of newly formed viral particles. Collectively, E1, E2, and E4 drive integration, replication, and transcription of the viral genome (Bhattacharrjee et al., 2021; Della Fera et al., 2021; Evande et al., 2023). E5 is involved in membrane signaling, cell proliferation, apoptosis, oncogenesis, and angiogenesis (Basukala and Banks, 2021; Estêvão et al., 2019). E6 and E7 are potent oncogenic proteins that interact with a wide array of cellular proteins. Their activity is essential for the development and maintenance of the malignant phenotype. Impairment of these proteins, or the inhibition of their gene expression, induces cellular senescence and growth arrest in HPV-positive cancer cells (Basukala and Banks, 2021; Estêvão et al., 2019). The principal function of E6 is its interaction with the tumor suppressor p53 (Martinez-Zapien et al., 2016). This interaction inhibits apoptosis, thereby allowing cells harboring genomic damage to evade cell death and continue proliferating. In addition, E6 engages with other host proteins, contributing to carcinogenic phenotypes, cellular immortalization, and cell invasion (Panczyszyn et al., 2018). E7 promotes degradation of the tumor suppressor retinoblastoma (pRB), leading abnormal centrosome synthesis and aneuploidy, ultimately resulting in genomic instability (Duensing et al., 2001). In addition, E7 interacts with other cellular targets to promote deregulation of cell proliferation and increased invasive potential. E6 and E7, acting independently or in concert, modulate a wide range of cellular pathways critical for cancer progression. These include immune evasion, dysregulation of cellular energetics, inhibition of growth suppressor pathways, sustained proliferative signaling, epigenetic reprogramming, metastasis, tumor-associated inflammation, and angiogenesis (Basukala and Banks, 2021; Estêvão et al., 2019).
L1 and L2 are structural proteins that constitute the capsomers of the viral capsid. Indeed, current vaccines against HPV are designed to elicit immune responses primarily targeting the strong antigenicity of L1 (Tsakogiiannis et al., 2022). In addition, the L1 gene sequence is used for HPV genotyping, while L2 plays a role in the intracellular transport of viral DNA to the nucleus of infected cells (Doorbar et al., 2015; Graham, 2017; Rosendo-Chalma et al., 2024). In the following sections, we review the mechanisms by which viral proteins are synthesized from a myriad of polycistronic mRNAs.
HR-HPVs proteins are synthesized using different mechanisms to initiate translation
HR-HPV proteins are expressed by polycistronic mRNAs transcribed from the different promoters in the long control region (LCR), the early (E) region and the late (L) region of the HPV-16 and HPV-18 genomes, respectively (Figure 2A) (Doorbar et al., 2015; Graham, 2017; Rosendo-Chalma et al., 2024). Overall, the high complexity of alternative splicing events of viral transcripts produces a plethora of mRNAs with diverse structures. In all mRNAs, the first ORF encodes E6. Full-length E6 is only translated from unspliced transcripts (De la Cruz-Hernández et al., 2005; Mesplede et al., 2012; Moral-Hernández et al., 2010; Schneider-Gädicke and Schwarz, 1986; Smotkin and Wettstein, 1986; Stacey et al., 1995; Toots et al., 2014). In addition, shorter versions of E6 termed E6I, E6II, E6III, E6IV, E6V, E6VI, E6^E7, E6^E7I, and E6^E7II are also expressed from alternative splicing events of the same mRNA (Ajiro and Zheng, 2015; Brant et al., 2018; Olmedo-Nieva et al., 2018). E7 is synthesized from several spliced transcripts and from unspliced mRNAs (Doorbar et al., 1990; Durst et al., 1991; Mesplede et al., 2012; Moral-Hernández et al., 2010; Sherman et al., 1992; Smotkin et al., 1989; Tang et al., 2006; Thierry et al., 1987). Moreover, multiple alternatively spliced transcripts and clusters of heterogeneous mRNAs expressed from different transcription start sites have also been identified in cultured cells, raff cultures, and tumorigenic tissues that only contain either E6 or E7 as the sole cistron (Braunstein et al., 1999; Glahder et al., 2003; Grassmann et al., 1996; Rosenstierne et al., 2003; Schneider-Gädicke and Schwarz, 1986; Smotkin and Wettstein, 1986). Figure 2B shows most of the multiple, polycistronic mRNAs transcribed from both the episomal and the integrated HR-HPVs genome (Graham, 2010; Graham, 2017; Wang et al., 2025).
*Different mechanisms drive the translation of a plethora of polycistronic mRNAs to synthesize the viral HPV proteins. (A) Structure of HR-HPV 16 and 18 genomic DNA. (B) Some mRNAs originated from episomal genome and from viral DNA integrated into the cellular genome. Thin arrows represent the promoters P
97 for HPV-16 and P
102 and P
105 for HPV-18; pAE, early polyadenylation signals; LCR, long control region; Squares represent exons; lines into mRNAs represent intercistronic regions. R.S., ribosome shunting; T.R., termination-reinitiation; L.S., leaky scanning; C.S., canonical scanning. E5, L1, and L2 cistrons might be translated from hypothetical IRES (thick gray arrows with a question mark). Adapted from Yu et al. (2022), Wang et al. (2025), García et al. (2024), Wang et al. (2025), Yu et al. (2022). A fuller list of the HPVs transcripts is described in the Papilloma Virus Episteme (PaVE) (Doomer et al., 2024; Van Doorslaer et al., 2017) at https://pave.niaid.nih.gov/#explore/transcript_maps and references therein.*
Surprisingly, In cultured cells and epithelial rafts the 5′-UTR regions of E6,E7 mRNAs are 7-9 nucleotides in length for HPV-16 (Stacey et al., 2000), and in HPV-18 the most represented transcripts possess either 3 nucleotides long 5′-UTR or completely lack it, i.e., the transcription starts at the A of the AUG codon that initiates translation (García et al., 2021; Romanczuk et al., 1990; Schneider-Gädicke and Schwarz, 1986; Smotkin and Wettstein, 1986; Thierry et al., 1987; Wang et al., 2011). Thus, the structure of viral mRNAs contrasts drastically with that of mRNAs across eukaryotes (both cellular and viral), with a 5′-UTR length average of 53–164 nucleotides (Leppek et al., 2018; Ryczek et al., 2023). In multicellular species, short 5′-UTRs have only been found in the human nuclear-encoded mRNAs harboring the element Translation Initiation of Short 5′-UTR (TISU), consisting of 12–30 nucleotide-long 5′-UTRs (Elfakess and Dikstein, 2008; Elfakess et al., 2011), and some mitochondrial mRNAs with 5′-UTRs shorter than five nucleotides (Gandin et al., 2016). Recently, we demonstrated that in cervical tumors E6,E7 mRNAs from HR HPV-18, HPV-39 and HPV-45 possess 5′-UTRs of 0–5 nucleotides which drive the translation of full-length E6 (García et al., 2021). For HPV-18, E6 is translated in a scanning-independent manner and is dependent on the RNA motif within the E6 coding region termed TILM, that drives the translation of E6 (García et al., 2021). However, the proteins that regulate E6 synthesis via the TILM motif in vivo are unknown. In contrast, the HPV-16 E6 cistron with a 7-9 nucleotides-long 5′-UTR is translated by the canonical mechanism (Stacey et al., 2000).
The synthesis of polycistronic mRNAs raises the fundamental question of how E1, E2, E4, E5, and E7 are translated downstream of E6. Evidence from cervical cancer cell lines suggest that the HPV-16 and HPV-18 E7 cistron is translated from spliced E6*I mRNA via translation termination-reinitiation (Tang et al., 2006). In a cell-free coupled in vitro transcription/translation system using rabbit reticulocyte lysates and a synthetic HPV-16 E6,E7 bicistronic mRNA, Tan et al. showed that full-length E6 translation also promotes termination-reinitiation on the E7 cistron AUG 3 to 6 nucleotides downstream the E6 stop codon (Tan et al., 1994).
The translation HPV-18 E1 has been studied by plasmid transient transfections of the green monkey kidney COS-7 fibroblast-like cultured cells (Remm et al., 1999). The distance between the E7 and E1 ORF consists of 6 nucleotides, and Remm et al. (1999) demonstrated that the translation of the E1 cistron downstream of E7 occurs by ribosome shunting. This group demonstrated that the translation of the E1 ORF is inhibited from monocistronic transcript that might arise from spurious transcription sites or partial degradation, and its translation is strongly favored from polycistronic rather than monocistronic mRNAs as a means to ensure E1 premature synthesis in early stages of the viral infection cycle.
The translation of HPV-16 E2 ORF has been studied in both COS cells and cell-free in vitro translation systems. In the transcripts E6I,E7,E2,E5, the E7 stop codon and the E2 AUG are separated by 33 nucleotides and the termination-reinitiation mechanism could translate E2 after E7 is translated. On the other hand, in the E6IV transcript, the E2 AUG is out of frame and upstream of the E6IV termination codon. Moreover, E2 AUG has a stronger Kozak motif than E6VI AUG. Thus, avoiding the upstream E6*IV AUG leaky scanning could also be the mechanism to initiate E2 translation (Alloul and Sherman, 1999).
HPV-16 E5 translation has been studied only in the mouse fibroblast NIH3T3 cells (Johnsen et al., 1995). HPV-16 and HPV-18 E1^E4, E5 mRNA is the most abundant transcript found in all cervical neoplasias (Sherman and Alloul, 1992; Stoler et al., 1992). However, HPV-16 E5 is not translated from this mRNA because it is rich in AUG triplets upstream the initiator AUG codon. In contrast, E2 and E5 are well expressed from the E2,E5 transcripts lacking the upstream E^E4 cistron, suggesting a synchronized, positive translational regulation of both E2,E5 ORFs (Johnsen et al., 1995). However, the mechanisms of E5 translation has not been elucidated. We have proposed the possible existence of IRES to drive the expression of E5 ORF (García et al., 2024) (represented in Figure 2B by gray arrows). However, experimental analyses should be performed to prove it.
HPV-18 E7 in COS transfected cells E7 is translated by the canonical mechanism that might result from the translation of a subset of short mRNAs originated from the P_105_ early promoter lacking the first nucleotides of E6 ORF (Remm et al., 1999). HPV-16 E7 translation was also studied in cell-free rabbit in vitro translation lysates and plasmid transfection of HeLa cells (Stacey et al., 1995; Stacey et al., 2000). In contrast, it was found that E7 ORF is translated by an exceptionally pervasive leaky scanning process in which the ribosomal initiation complex does not recognize the AUG start codon of the upstream E6 cistron, and scans through the mRNA over 13 upstream AUG codons until it reaches the E7 ORF TIS (Stacey et al., 1995; Stacey et al., 2000).
L1 and L2 are transcribed from the late region of the HR-HPVs genome (Figure 2C) (Doorbar et al., 2015; Graham, 2017; Nelson and Mirabello, 2023; Rosendo-Chalma et al., 2024). So far, no mechanism of mRNA translation has been elucidated for the L1 and L2 mRNAs, but we propose here that they might be translated by IRESs. Further experimental analyses also should be performed to prove it. In Table 1 we summarize the mechanisms used to translate the different HPV-16 and HPV-18 cistrons.
Codon usage regulates viral mRNA translation
In all organisms, most amino acids are encoded by multiple synonymous codons. However, codon usage is not uniform across the different species. The preference for specific codons over others during mRNA translation is referred to as codon bias or codon preference (Parvathy et al., 2022). Due to the absence of their own translational machinery, viruses have evolved to optimize their codon usage to match that of their host. By aligning their codon usage patterns with the host’s tRNA pool, viruses enhance the translational efficiency of their own proteins (Kaleem et al., 2025; Simón et al., 2021).
Viruses often employ suboptimal codon usage to reduce the translation rate of viral proteins as a strategy to evade host immune surveillance and response. This involves the use of codons that are less favorable for translation (Kaleem et al., 2025; Mordstein et al., 2021). Indeed, in a wide range of HPV types, codon bias shows poor alignment with the average human codon usage preferences (Cladel et al., 2010; Félez-Sánchez et al., 2015; Ren et al., 2025; Zhao and Chen, 2011). Moreover, a study of the codon usage bias across 79 HPVs showed strong differences among the eight cistrons encoding viral proteins toward 18 codons with T at the third position. Among them, L2 ORFs showed the highest codon bias and E4 the lowest (Zhao et al., 2003). The codon bias of E6,E7 mRNAs poorly matches the preferred codons by host cells, which strongly diminishes the rate of protein translation. As expected, changing the rare toward optimal codons of human cells (Müller, 2005) or the co-transfection of different cultured cells with a plasmid expressing tRNA^Ser(CGA)^ (Gu et al., 2004) enhanced protein synthesis of the viral proteins. Transfection of a codon-optimized HPV-16 E6 gene also increased protein expression in cultured human cell (Lin et al., 2006; Samorski et al., 2006). Regarding E7, supplementation of rat liver tRNAs to the pool of a cell-free in vitro translation system from rabbit reticulocyte (De Pasquale and Kanduc, 1998), or the use of a codon-optimized plasmid transfected into human cells (Steinberg et al., 2005) significantly enhanced E7 synthesis. It was further observed that the codon bias of HPV genes couples viral gene expression with epithelium differentiation throughout virus life cycle and carcinogenesis (Gu et al., 2007; Zhao et al., 2005; Zhou et al., 1999).
The study of codon usage bias has been fundamental to the design of vaccines against HR-HPVs. In the two widely used commercial vaccines, Gardasil and Cervarix, codon optimization was employed to enhance the production of virus-like particles (VLPs) derived from the L1 protein, which serves as the primary immunogenic target (Kaleem et al., 2025; Markowitz and Schiller, 2021).
Integrated HPVs produce chimeric E6, E7 mRNAs fused with cellular genes
The life-cycle of mRNAs is tightly controlled by numerous cis-regulatory elements located at the 5′- and 3′-UTRs that control the translation and transport of the transcripts, are target of nucleases, or bind to a myriad of regulatory molecules (Leppek et al., 2018; Mayr, 2017; Ryczek et al., 2023). Consequently, mutations in the mRNA UTRs may lead to the uncontrolled expression of a wide variety of mRNAs and, often trigger the development of diverse diseases (Chatterjee and Pal, 2009), including cancer (Chan et al., 2023; Huang et al., 2022; Li and Lu, 2013; Schuster and Hsieh, 2019; Silanes et al., 2007; Zhang et al., 2022).
The regulatory elements that control polyadenylation, degradation, localization, and transport of mRNAs are mostly located in the 3′-UTR (Mayr, 2017). In most tumors, HPV DNA is generally integrated into the genome of epithelial cells, unlike productive infections where is found in episomal form (Figure 3). The viral genome integration is a stochastic process over almost all chromosomes. During this, some viral genomes may be integrated into cancer-related genes which may promote tumorigenesis. Moreover, as a consequence of this integration, the E6 and E7 genes lose their natural 3′-UTR, disrupt the viral E2 ORF, and gain a cellular DNA fragment that is transcribed as a viral-cellular chimeric 3′-UTR (Figure 3). The promoter controlling E6 and E7 genes contains binding sites for E2 protein and binding of E2 represses E6, E7 mRNA transcription. Therefore, loss of the E2 cistron leads to an increase in the E6, E7 mRNA transcription and protein synthesis (Jeon et al., 1995; Lace et al., 2011; Vinther et al., 2005; Wentzensen et al., 2002; Wentzensen et al., 2004). Thus, the main consequence of HPV genome integration is the upregulation and stabilization of E6,E7 mRNAs, leading to the E6 and E7 overexpression and the promotion of carcinogenesis (Couturier et al., 1991; Durst et al., 1987; Ehrig et al., 2020; Jeon and Lambert, 1995; Wentzensen et al., 2002; Ziegert et al., 2003).
*mRNA expression patterns from integrated (fusion) and episomal HPV 16/18 DNA. Diagram illustrating the characteristic structure of viral mRNAs produced from intact episomal genomes (left) or the viral DNA integration into the cellular genome (right). Only the cistrons (colored boxes) controlled by the long control region (LCR, gray box) is depicted. The 3′-UTR of these transcripts is shown. Colored boxes, viral cistrons; m
7
G, mRNA cap structure; AAAA, mRNa poly(A) tail.*
The viral genome integration into the cell genome plays a critical role in triggering carcinogenesis and is one of the molecular features that best defines the transition to carcinogenic transformation (Groves and Coleman, 2015; Groves and Coleman, 2018; Wentzensen et al., 2004). Although the stability of chimeric mRNAs increases (Ehrig et al., 2020; Jeon and Lambert, 1995; Vinther et al., 2005), whether the translation rate of the viral-cellular fusion mRNAs also increases is unknown and deserves further experimental analysis. In Table 2 we summarize the diverse events of viral-cellular fusion transcripts reported in different cells or tissues that generate mRNAs possessing chimeric 3′-UTRs and the consequences in cancer.
Old questions, new insights
Cervical cancer remains a significant global public health challenge. Despite substantial advances in understanding the translation of HPV mRNAs, several longstanding questions remain unresolved. Among them is the mechanism by which the E5, L1, and L2 cistrons are translated. We speculate that these cistrons may utilize internal ribosome entry sites (IRESs); however, this hypothesis requires experimental validation. For certain viral proteins, such as E7 and E2, alternative translation mechanisms have been proposed. Therefore, further studies are necessary to clarify these observations. Additionally, the impact of cellular fragment fusion on the translation of chimeric virus–host mRNAs remains largely unexplored.
To date, most studies investigating the translation of HPV proteins have been conducted in heterologous systems—such as monkey COS-7 cells, mouse NIH3T3 cells, or rabbit reticulocyte lysates. Therefore, these findings must be validated in human cervical tissues, particularly throughout the process of epithelial differentiation during carcinogenesis.
Understanding the translation mechanisms of viral mRNAs is crucial for the future development of targeted therapies for cervical cancer. This will be instrumental in designing therapeutic strategies that combine translation inhibitors with agents directly targeting viral oncoproteins. Recent findings have highlighted promising pharmacological approaches. Two molecules targeting eIF4E, a key translation initiation factor, are currently being investigated for the treatment of HPV-associated cancers. Cercosporamide has been shown to reduce chemotherapy-induced phosphorylated eIF4E (p-eIF4E) levels, which are implicated in cancer progression, in mouse tumor models (Zhu et al., 2021). Ribavirin, another agent targeting p-eIF4E, has undergone clinical evaluation for HPV-related malignancies. Oral administration of ribavirin resulted in decreased p-eIF4E levels and was well-tolerated (Burman et al., 2022) (clinical trial NCT02308241; ClinicalTrials.gov).
Finally, several drugs are currently undergoing testing in animal models or clinical trials targeting the translation machinery across various cancers (García et al., 2024). These research efforts will be pivotal for the future development of cervical cancer therapies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ajiro M.Zheng Z. M. (2015). E 6^E 7, a novel splice isoform protein of human papillomavirus 16, stabilizes viral E 6 and E 7 oncoproteins via HSP 90 and GRP 78. m Bio 6, e 02068–e 02014. 10.1128/m Bio.02068-14 25691589 PMC 4337564 · doi ↗ · pubmed ↗
- 2Alloul N.Sherman L. (1999). The E 2 protein of human papillomavirus type 16 is translated from a variety of differentially spliced polycistroniic m RN As. J. Gen. Virol. 80, 29–37. 10.1099/0022-1317-80-1-29 9934680 · doi ↗ · pubmed ↗
- 3Alteri R.Baptiste D.Bell E. B. (2025). Cancer prevention and early detection. Facts and figures 2025-2026. American Cancer Society. 860025.
- 4Basukala O.Banks L. (2021). The not-so-good, the bad and the ugly: HPV E 5, E 6 and E 7 oncoproteins in the orchestration of carcinogenesis. Viruses 13, 1892. 10.3390/v 13101892 34696321 PMC 8541208 · doi ↗ · pubmed ↗
- 5Bhattacharrjee R.Das S. S.Biswal S. S.Nath A.Das D.Basu A. (2021). Mechanistic role of HPV-associated early proteins in cervical cancer: molecular pathways and targeted therapeutic strategies. Crit. Rev. Oncol. Hematol. 174, 103675. 10.1016/j.critrevonc.2022.103675 35381343 · doi ↗ · pubmed ↗
- 6Brant A. C.Menezes A. N.Felix S. P.de Almeida L. M.Sammeth M.Moreira M. A. M. (2018). Characterization of HPV integration, viral gene expression and E 6E 7 alternative transcripts by RNA-Seq: a descriptive study in invasive cervical cancer. Genomics 111, 1853–1861. 10.1016/j.ygeno.2018.12.008 30552977 · doi ↗ · pubmed ↗
- 7Braunstein T. H.Madsen B. S.Gavnholt B.Rosenstierne M. W.Koefeld Johnsen C.Norrild B. (1999). Identification of a new promoter in the early region of the human papillomavirus type 16 genome. J. Gen. Virol. 80, 3241–3250. 10.1099/0022-1317-80-12-3241 10567657 · doi ↗ · pubmed ↗
- 8Brito Querido J.Días-López I.Ramakrishnan V. (2024). The molecular basis of translation initiation and its regulation in eukaryotes. Nat. Rev. Mol. Cell. Biol. 25, 168–186. 10.1038/s 41580-023-00624-9 38052923 · doi ↗ · pubmed ↗