Mitochondrial diabetes: From suspicion in primary care to a multidisciplinary family approach
Blanca Adriana Macías Martínez, Alba Dalmau Vila, Marta Hernández García, Maria Antonia Lafarga Giribets

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
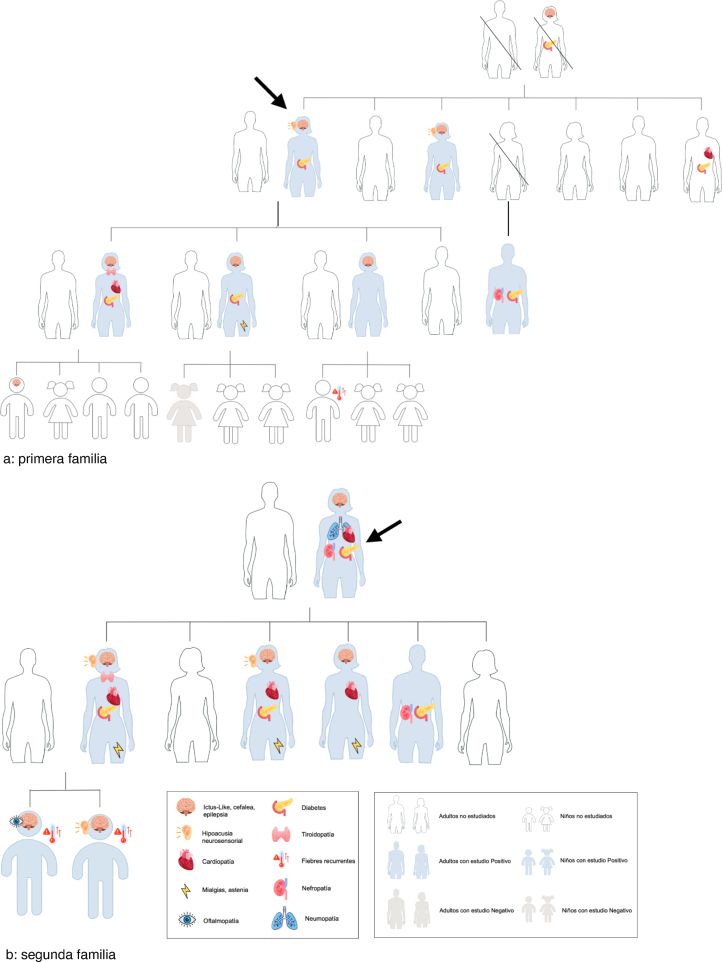 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMitochondrial Function and Pathology · Metabolism and Genetic Disorders · Diabetes and associated disorders
La mutación del DNA mitocondrial (mtDNA) es una de las causas más frecuentes de enfermedades hereditarias1, con tres particularidades: a) herencia principalmente por vía materna; b) la mutación patogénica puede afectar en diversa proporción a los diferentes tejidos, lo que se conoce como heteroplasmia, y dificulta el diagnóstico, y c) las copias de mtDNA varían según el tejido y a lo largo del tiempo1.
La prevalencia de enfermedades mitocondriales varia de 1/5.000 niños hasta 1/8.000 adultos2. La mutación 3243A>G en el gen MT-TL1 es la más frecuente3. Principalmente se manifiesta en órganos con alto consumo de energía, como el sistema nervioso, corazón y páncreas, aunque puede afectar prácticamente a cualquier tejido, sistema y órgano4. El fenotipo más común (30%) es el síndrome MIDD (diabetes mellitus [DM] de herencia materna con hipoacusia neurosensorial) y un 10% se puede presentar como MELAS (miopatía mitocondrial, encefalopatía, acidosis láctica y episodios que simulan ictus), acompañado o no de DM3. Se ha acuñado el término «portador latente» para aquellos pacientes con mutación mitocondrial pero sin síntomas clínicos3.
El MIDD es el fenotipo más común del sistema endocrino, y el 85% de los pacientes presentan la mutación A32434. La DM suele iniciarse de manera similar a la DM2 pero en edades más jóvenes (30-40 años de media). Puede ser tratada inicialmente con dieta y sulfonilureas. La metformina está contraindicada por riesgo de acidosis láctica. Generalmente presentan un declive rápido de la función de células β y necesitan insulina en 2-4 años, dependiendo de los niveles de heteroplasmia. Los pacientes suelen presentar un índice de masa corporal (IMC) normal/bajo4, 5.
En las enfermedades mitocondriales el diagnóstico requiere un alto grado de sospecha. El hecho de que un paciente presente DM2 con IMC normal e hipoacusia es un factor clave para derivarlo a un servicio especializado3. El diagnóstico se establece con la secuenciación de mtDNA en sangre u orina, aunque en ocasiones es necesario realizar una biopsia de músculo esquelético6 con biopsia de piel. Una vez localizado el caso índice, se debe extender el estudio a familiares por rama materna.
No existe una terapia curativa por el momento, aunque actualmente se realizan diversos ensayos: combinado de vitaminas, antioxidantes y cofactores, modificaciones adicionales basadas en la etiología genética, fenotipos y hallazgos bioquímicos. A las mujeres portadoras de la mutación mitocondrial se les ofrece consejo genético y opciones reproductivas6.
Favorecido por la consanguinidad, existen familias estudiadas en Países Bajos, Japón, Australia y Turquía4, 5 afectas por dichos síndromes.
En nuestra región recogimos los casos de dos familias con alta penetración de síndrome MELAS y MIDD, en los que el diagnostico se realizó por la alta sospecha del caso índice con diabetes y sordera. El probando de la primera familia (fig. 1a) presentaba DM desde los 23 años, insulinotratada y con mal control, además de episodios ictus-like, DM gestacional previa y fibrilación auricular. El caso índice de la familia 2 (fig. 1b) presentaba DM insulinotratada desde los 32 años, DM gestacional previa y síntomas digestivos, neumológicos, cardiológicos y tiroideos. Estudiados los familiares, destaca la alta penetrancia de la mutación. Los pacientes familiares presentaban, además de DM, taquicardias/arritmias, déficit intelectual, pérdida de agudeza visual, mialgias, hipotiroidismo, anemia ferropénica y retrasos madurativos en el caso de los niños.Figura 1Genograma de los pacientes estudiados. a) primera familia; b) segunda familia.
Se necesita, pues, un alto índice de sospecha por parte del médico de familia para diagnosticar esta entidad. La sospecharemos en familias con pacientes diagnosticados de DM2 relativamente jóvenes (20-60 años), con herencia por vía materna, sin síndrome metabólico e IMC dentro de la normalidad, con otras alteraciones asociadas como hipoacusia, miopatía, neuropatía óptica y/o crisis comiciales. El hecho de padecer una DM gestacional en pacientes sin obesidad/síndrome metabólico también es relevante en nuestras familias.
Financiación
Este estudio no recibió ningún tipo de financiación.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Consideraciones éticas
Los autores declaran que se han seguido los protocolos establecidos por sus respectivos centros sanitarios para acceder a los datos de las historias clínicas a los fines de poder realizar este tipo de publicación con finalidad de investigación/divulgación para la comunidad científica.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chinnery P.F.Elliott H.R.Hudson G.Samuels D.C.Relton C.L.Epigenetics, epidemiology and mitochondrial DNA diseases Int J Epidemiol.4120121771872228713610.1093/ije/dyr 232PMC 3304530 · doi ↗ · pubmed ↗
- 2Aldossary A.M.Tawfik E.A.Alomary M.N.Alsudir S.A.Alfahad A.J.Alshehri A.A.Recent advances in mitochondrial diseases: From molecular insights to therapeutic perspectives Saudi Pharm J.302022106510783616457510.1016/j.jsps.2022.05.011PMC 9508646 · doi ↗ · pubmed ↗
- 3De Laat P.Koene S.van den Heuvel L.P.W.J.Rodenburg R.J.T.Janssen M.C.H.Smeitink J.A.M.Clinical features and heteroplasmy in blood, urine and saliva in 34 Dutch families carrying the m.3243 A >G mutation J Inherit Metab Dis.352012105910692240301610.1007/s 10545-012-9465-2PMC 3470685 · doi ↗ · pubmed ↗
- 4Li D.Liang C.Zhang T.Marley J.L.Zou W.Lian M.Pathogenic mitochondrial DNA 3243 A>G mutation: From genetics to phenotype Front Genet.13202295118510.3389/fgene.2022.951185 PMC 958266036276941 · doi ↗ · pubmed ↗
- 5Maassen J.A.’t Hart L.M.van Essen E.Heine R.J.Nijpels G.Tafrechi R.S.J.Mitochondrial diabetes: Molecular mechanisms and clinical presentation Diabetes.53Suppl 1200410310910.2337/diabetes.53.2007.s 10314749274 · doi ↗ · pubmed ↗
- 6Gorman G.S.Chinnery P.F.Di Mauro S.Hirano M.Koga Y.Mc Farland R.Mitochondrial diseases Nat Rev Dis Primers.22016160802777573010.1038/nrdp.2016.80 · doi ↗ · pubmed ↗