The application of desymmetric enantioselective reduction of cyclic 1,3-dicarbonyl compounds in the total synthesis of terpenoid and alkaloid natural products
Dong-Xing Tan, Fu-She Han

TL;DR
This review discusses how a specific chemical reaction helps in making complex natural products like terpenoids and alkaloids by creating chiral building blocks.
Contribution
The paper summarizes recent advances in using desymmetric enantioselective reduction for synthesizing terpenoids and alkaloids from 2016 to 2025.
Findings
Desymmetric enantioselective reduction is effective for creating complex ring systems with multiple stereocenters.
Five-membered cyclic 1,3-dicarbonyl compounds are widely used in the synthesis of terpenoids and alkaloids.
Six-membered cyclic 1,3-dicarbonyl compounds have also been applied in the synthesis of some terpenoids.
Abstract
The desymmetric enantioselective reduction of cyclic 1,3-dicarbonyl compounds is a powerful tool for the construction of ring systems bearing multiple stereocenters including all-carbon quaternary stereocenters, which are widely useful chiral building blocks for the total synthesis of structurally complex natural products. On the other hand, terpenoids and alkaloids, with their intricate and diverse skeletal frameworks as well as the broad range of biological activities, have long been a major focus for synthetic chemists. Over the past fifteen years, significant progress has been made in the total synthesis of complex terpenoid and alkaloid natural products by strategically applying desymmetric enantioselective reduction. Advance before 2016 in this area has been overviewed in an elegant review article. Since then, a series of more challenging terpenoid and alkaloid natural products…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
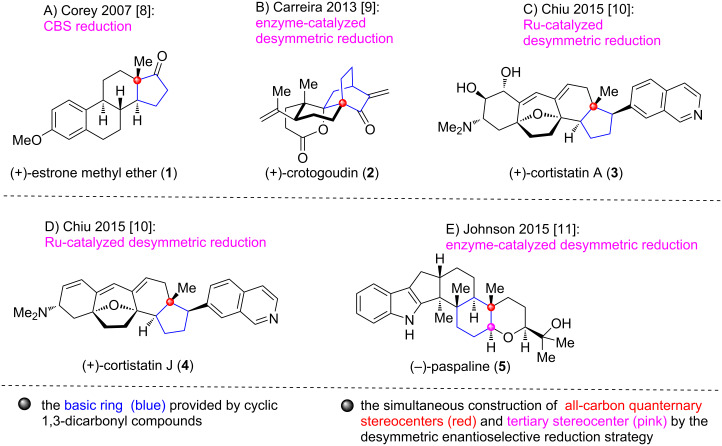 Figure 1
Figure 1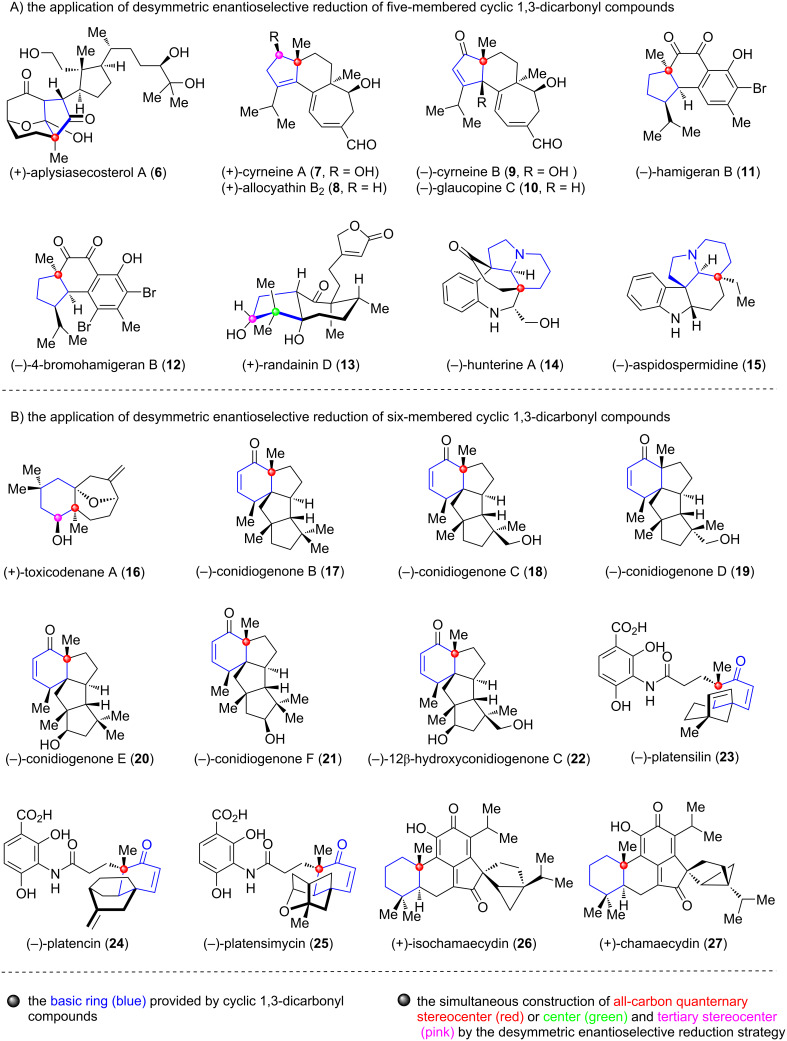 Figure 2
Figure 2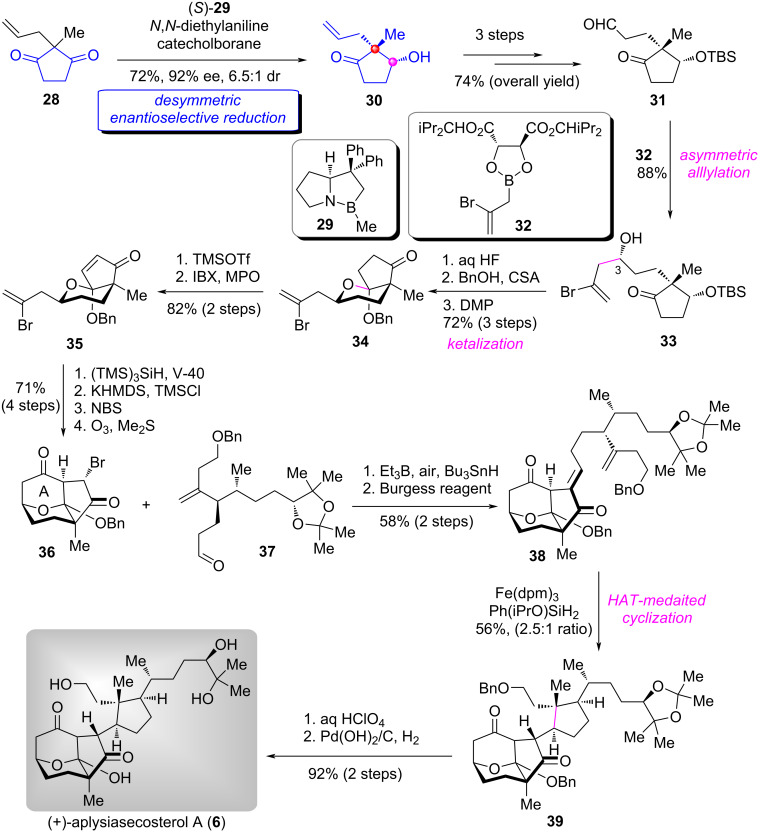 Scheme 1
Scheme 1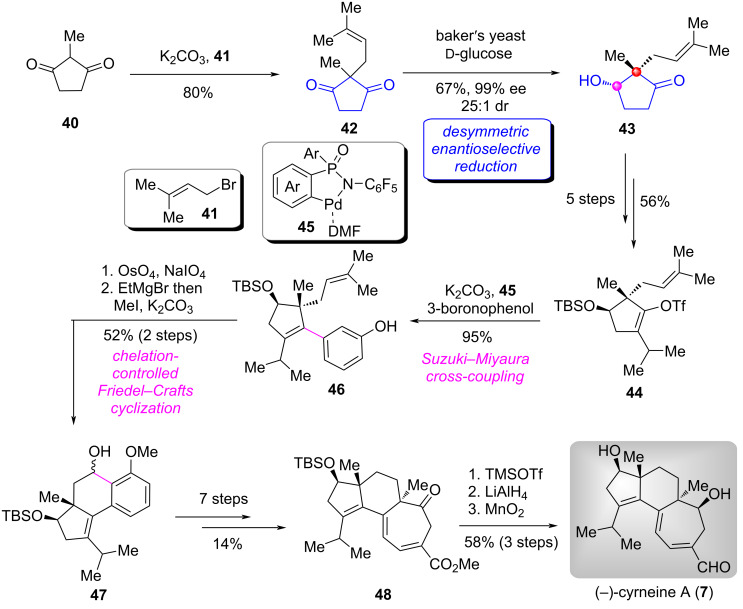 Scheme 2
Scheme 2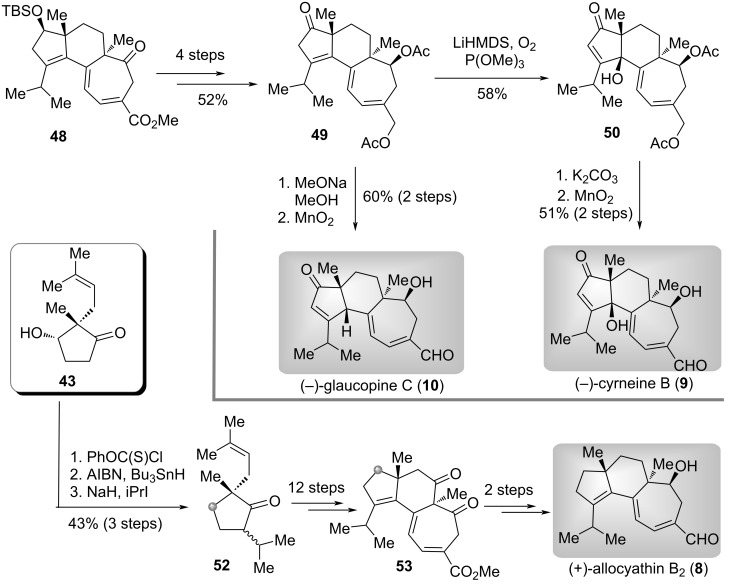 Scheme 3
Scheme 3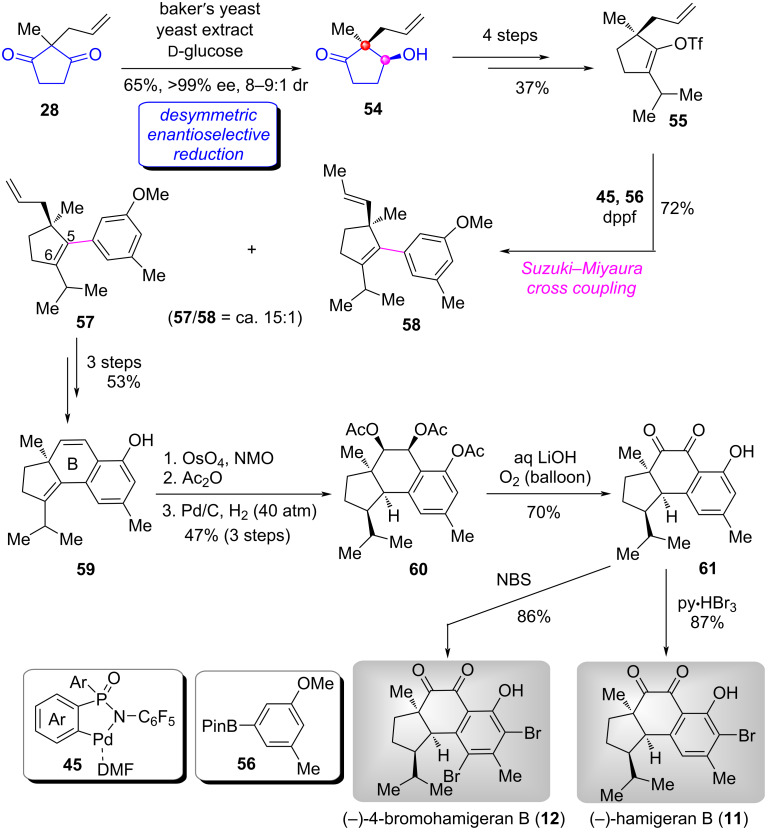 Scheme 4
Scheme 4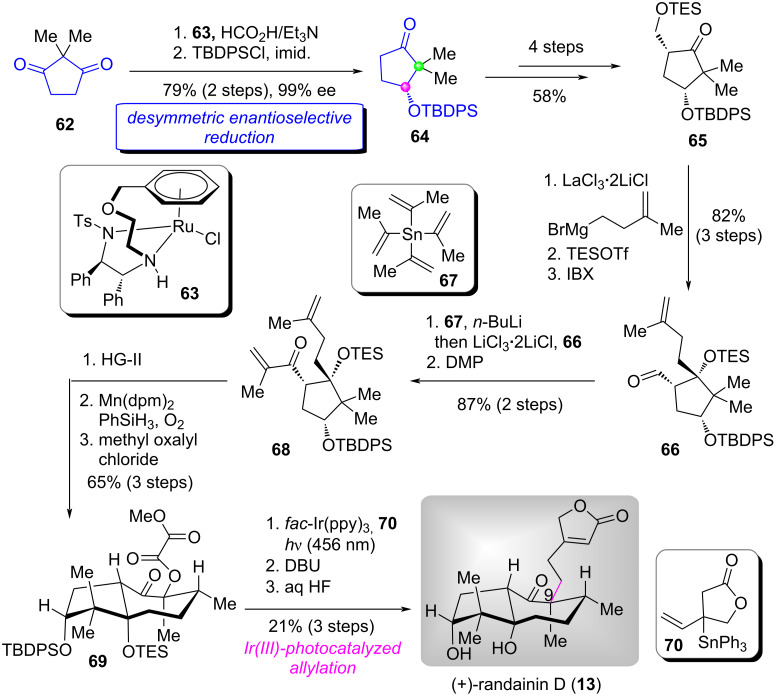 Scheme 5
Scheme 5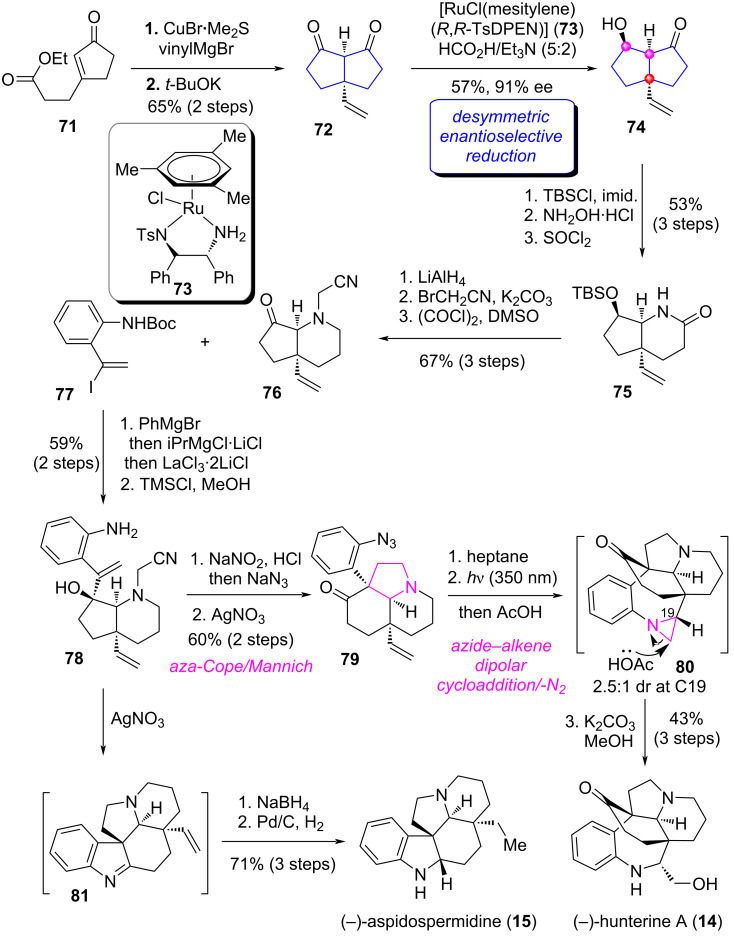 Scheme 6
Scheme 6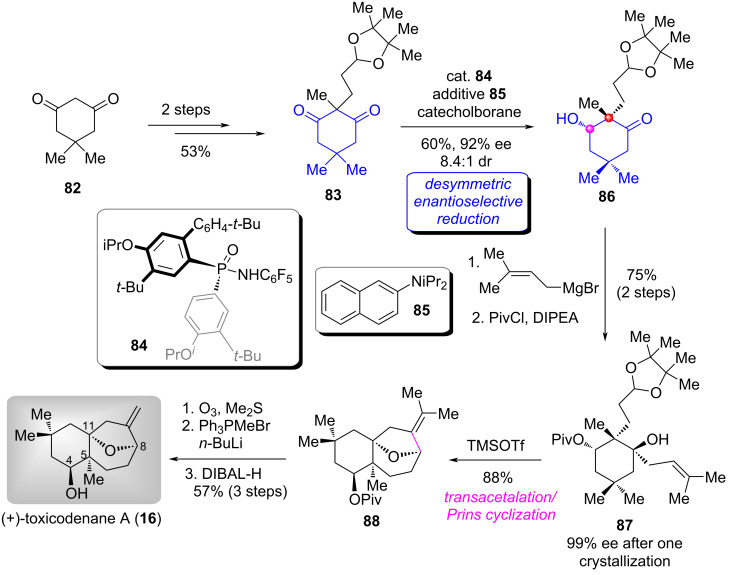 Scheme 7
Scheme 7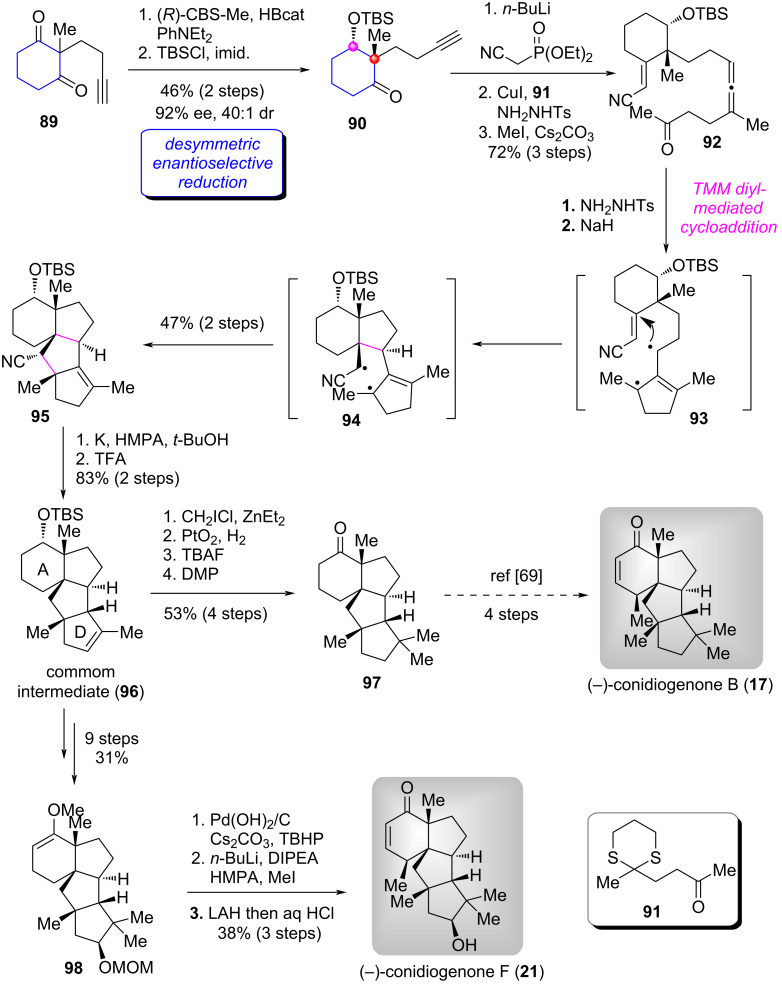 Scheme 8
Scheme 8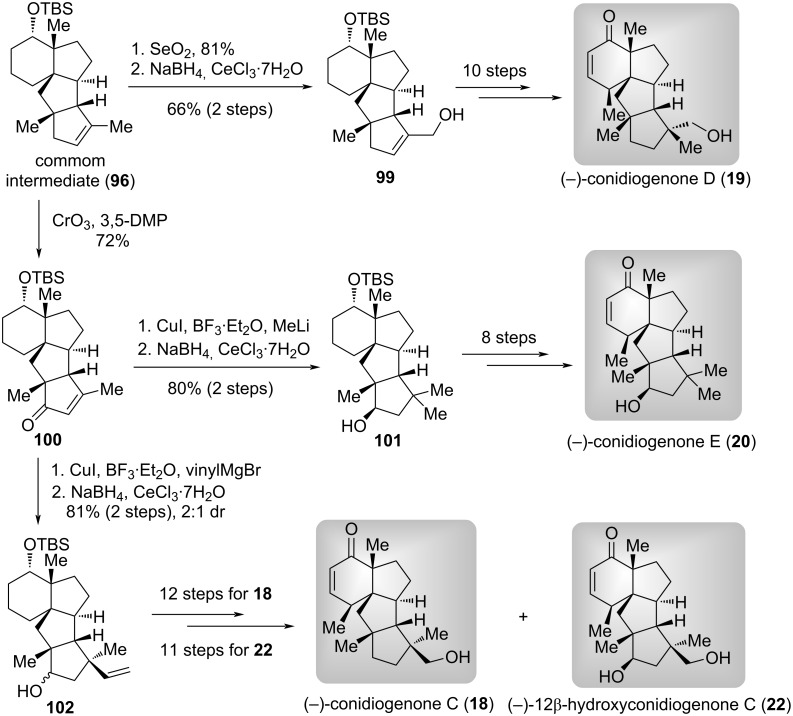 Scheme 9
Scheme 9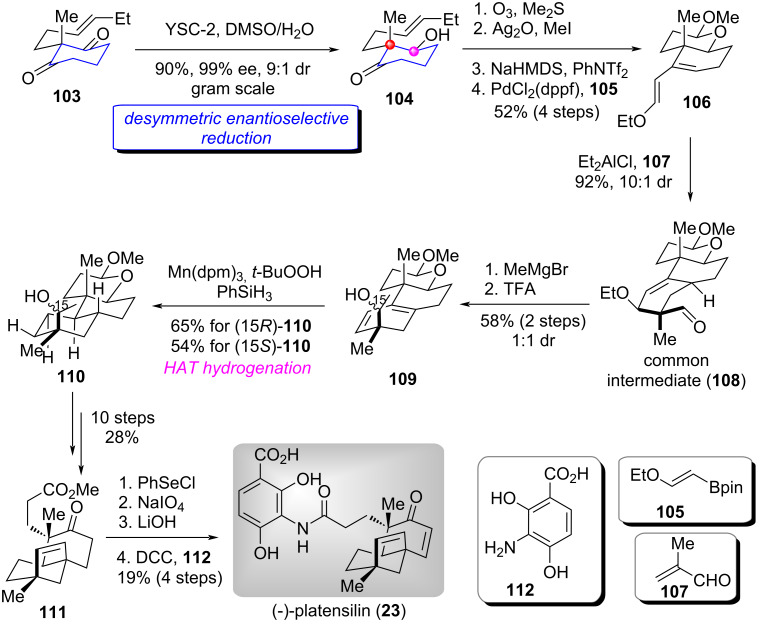 Scheme 10
Scheme 10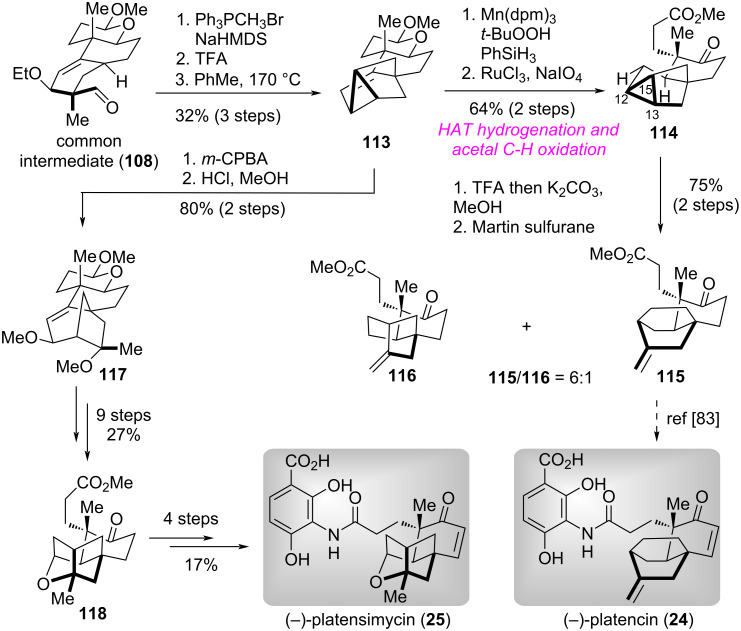 Scheme 11
Scheme 11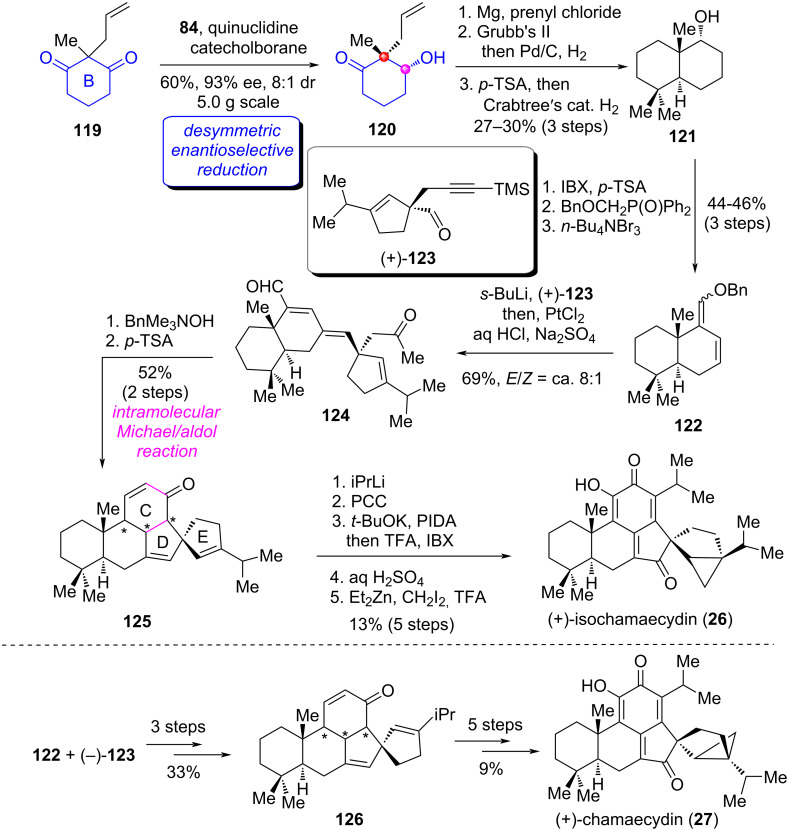 Scheme 12
Scheme 12Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced Synthetic Organic Chemistry · Alkaloids: synthesis and pharmacology · Asymmetric Synthesis and Catalysis
Introduction
Terpenoids and alkaloids are two major classes of highly important natural products because they usually exhibit diverse and important biological activities, such as antitumor, anti-inflammatory, and antiarrhythmic effects etc., and show potential to be developed into drug candidates or novel medications for treating human diseases [1–4]. However, their scarcity in nature limits further research into their biological activities. Total synthesis is an important device to address the shortage of natural product sources. Nevertheless, the asymmetric total synthesis of terpenoid and alkaloid natural products presents significant challenges due to their complex and diverse ring systems and the presence of multiple stereocenters, including all-carbon quaternary stereocenters. Consequently, the development of novel methods and strategies to achieve efficient asymmetric total synthesis of complex terpenoid and alkaloid natural products has drawn considerable attention from synthetic chemists.
Over the past decades, the development of desymmetric enantioselective reduction strategy of cyclic 1,3-dicarbonyl compounds has invigorated the field of terpenoid and alkaloid natural products synthesis because of its multiple advantages. Specifically, such strategy allows for an efficient construction of multiple chiral centers by employing easily accessible or commercially available symmetric cyclic prochiral dicarbonyl substrates. In addition, various approaches could be used for the desymmetrization reactions such as enzyme catalytic-, organocatalyst-, and transition-metal-catalyzed reductions [5–7]. Advance about the synthesis of several terpenoid and alkaloid natural products (1–5, Figure 1) [8–11] has been achieved in this area as indicated by an elegant review in 2016 [12], which will not be discussed in this review.
Several representative terpenoid and alkaloid natural products synthesized by applying desymmetric enantioselective reduction strategy of cyclic 1,3-dicarbonyl compounds before 2016.
Although only a limited number of terpenoids and alkaloids were synthesized by applying the desymmetric enantioselective reduction strategy of cyclic 1,3-dicarbonyl compounds before 2016, their success has provided a reference for the subsequent synthesis of other types of terpenoid and alkaloid natural products. In fact, over the past ten years, the number of terpenoid and alkaloid natural products synthesized utilizing this strategy has increased significantly compared to the period before 2016. However, there is currently a lack of systematic summary regarding the application of this strategy in the total synthesis of terpenoids and alkaloids over this time span. This review will provide a comprehensive overview of the recent advances (2016–2025), specially, we first summarize chronologically the application of desymmetric enantioselective reduction of five-membered cyclic 1,3-dicarbonyl compounds in the synthesis of terpenoid and alkaloid natural products 6–15 (Figure 2A). Subsequently, the advances in the utilization of six-membered cyclic 1,3-dicarbonyl compounds for the synthesis of terpenoid natural products 16–27 will be introduced in chronological order (Figure 2B). After that, a brief outlook of the desymmetric enantioselective reduction strategy of cyclic 1,3-dicarbonyl compounds in the total synthesis of natural products will be discussed. We hope this review can stimulate the methodology development and application of this strategy toward further improving the synthetic efficiency of natural product synthesis.
Selected terpenoid and alkaloid natural products synthesized by applying desymmetric enantioselective reduction strategy of cyclic 1,3-dicarbonyl compounds in 2016–2025.
Review
Total synthesis of terpenoid and alkaloid natural products through the desymmetric enantioselective reduction of five-membered cyclic 1,3-dicarbonyl compounds
Total synthesis of (+)-aplysiasecosterol A
(+)-Aplysiasecosterol A (6) is a secosteroid that was isolated from the sea hare Aplysia kurodai by Kita and Kigoshi in 2015 [13]. Due to its natural scarcity, the biological activity has not been explored. In 2018, Li and co-workers accomplished the first asymmetric total synthesis of (+)-aplysiasecosterol A (6) by employing a desymmetric enantioselective reduction strategy of 1,3-cyclopentanedione derivative as the key transformation [14].
Their synthesis features a highly efficient desymmetric enantioselective reduction of diketone 28 for preparing alcohol 30 under the CBS conditions [8] with (S)-29 as the catalyst (Scheme 1) [14]. Notably, this reaction could be performed on multiple gram scales with satisfactory yield (72%) and ee value (92%). Protection of the alcohol group in 30 with TBSCl followed by modification of the terminal double bond afforded ketoaldehyde 31. The 2-bromoallylation [15] of 31 with boronic ester 32 stereoselectively constructed the C3–OH group to give homoallylic alcohol 33. Next, a successive manipulation by removal of TBS group, CSA-catalyzed ketalization, and DMP oxidation of the secondary alcohol to a ketone allowed for rapid construction of bicyclic ketone 34 in high overall yield. The oxidative dehydrogenation of 34 gave α,β-unsaturated bicyclic ketone 35 smoothly. Sequential A-ring construction and functional group modifications of 35 produced the diketone 36. Subsequently, 36 and aldehyde 37, which was prepared with 9 steps from commercially available (+)-citronellol, underwent a Reformatsky-type radical addition under the conditions of Et_3_B/air/Bu_3_SnH to deliver aldol product [16]. Dehydration of the secondary alcohol gave (E)-38. The HAT radical cyclization [17] of 38 in the presence of Fe(dpm)3/Ph(iPrO)SiH_2_ proceeded smoothly to furnish the tetracyclic product 39 in 56% yield with 2.5:1 ratio. Finally, removal of the acetonide and the Bn protecting group completed the total synthesis of (+)-aplysiasecosterol A (6).
The total synthesis of (+)-aplysiasecosterol A (6) by Li [14].
Total synthesis of (+)-cyrneine A, (−)-cyrneine B, (−)-glaucopine C, and (+)-allocyathin B2
The cyrneine diterpenoids represent an important subfamily of cyathane diterpenoids which possess a common 5-6-7 fused tricarbocyclic core with two all-carbon quaternary stereocenters. Many impressive syntheses have been reported to date [18–30]. In 2018, the group of Han accomplished the total synthesis of (+)-cyrneine A (7), (−)-cyrneine B (9), (−)-glaucopine C (10), and (+)-allocyathin B_2_ (8) by a collective manner [31]. In their synthetic route, an enzyme-catalyzed desymmetric enantioselective reduction of 1,3-cyclopentanedione derivative was adopted as one of the key reactions, which facilitated the construction of the five-membered ring bearing an all-carbon quaternary center as the key chiral building block.
Their synthesis began with 1,3-dione 40 (Scheme 2) [31–32], allylation of this substrate with allylic bromide 41 afforded the 1,3-cyclopentanedione derivative 42. Next, the baker′s yeast-catalyzed desymmetric enantioselective reduction of 42 gave the α-hydroxyketone 43 in satisfactory yield and excellent stereoselectivity and diastereoselectivity on a decagram scale [33–34]. Functional group modifications and transformations of 43 produced hydroxyketone 44. Due to the steric hindrance of this substrate, the subsequent Suzuki cross coupling reaction with 3-boronophenol proceeded in low yield. To address this issue, Han′s group employed a novel palladacycle catalyst 45, previously developed by their group [35–37]. This catalytic system efficiently overcame the challenge and furnished the coupling product 46 in high yield. Oxidative cleavage of the double bond in 46 followed by Mg(II)-mediated chelation-controlled Friedel–Crafts cyclization delivered secondary alcohol 47, which was elaborated to ketone 48 via a seven-step transformation. Finally, removal of the TBS group in 48 followed by a sequential reduction and selective oxidation of allylic primary alcohol achieved the total synthesis of (−)-cyrneine A (7).
The total synthesis of (−)-cyrneine A by Han [31].
The authors then moved forward to the synthesis of other target molecules (Scheme 3) [31–32]. Oxidation state adjustment of 48 led to the ketone 49. Starting from this common intermediate, firstly, base-promoted double bond migration and oxidation at the γ-position gave tertiary alcohol 50. Deprotection of acetyl in 50 followed by selective oxidation delivered (−)-cyrneine B (9). Secondly, a based-mediated concomitant double bond migration and deacetylation, and selective oxidation of allylic primary alcohol accomplished the total synthesis of (−)-glaucopine C (10). On the other hand, Barton–McCombie deoxygenation and isopropylation of 43 produced ketone 52. Subsequently, by employing the same procedures for the synthesis of (−)-cyrneine A (7), the synthesis of (+)-allocyathin B_2_ (8) could also be achieved smoothly from 52 by utilizing diketone 53 as the intermediate. The diverse syntheses of these terpenoids enabled by the desymmetric enantioselective reduction of cyclic 1,3-dicarbonyl compounds demonstrated the significance of the desymmetric reduction strategy in the synthesis of structurally complex natural products.
The total syntheses of three cyrneine diterpenoids by Han [31–32].
Total synthesis of (−)-hamigeran B and (−)-4-bromohamigeran B
(−)-Hamigeran B (11) and (−)-4-bromohamigeran B (12) were isolated from the sponge Hamigera tarangaensis, and show cytotoxicity against various tumor cells. Notably, compound 11 exhibits 100% inhibition against herpes and polio viruses without significant host cell cytotoxicity [38]. Since their isolation, the synthesis of 11 and 12 have been reported by many groups [39–50]. For an efficient synthesis of these two natural products, Han and co-workers [51] adopted an alternative route utilizing the desymmetric enantioselective reduction strategy of a 1,3-cyclopentanedione derivative as the key transformation. Both (−)-hamigeran B (11) and (−)-4-bromohamigeran B (12) were synthesized by a divergent manner with a longest linear sequence of 14 steps from the known symmetrical diketone 28 (see Scheme 1) [14]. More importantly, this route allowed for the synthesis of target compounds at 100 mg scale in a single batch.
As shown in Scheme 4 [51], the enzyme-catalyzed desymmetric enantioselective reduction of 28, afforded hydroxyketone 54 in 65% yield with >99% ee and 8–9:1 dr on multigram scale [34]. Functional group transformations of 54 in four steps produced sterically hindered allyl triflate 55. By employing the palladacycle catalyst 45-catalyzed Suzuki cross-coupling reaction of sterically hindered substrates developed by Han [35–37], the coupled product 57 was obtained in a satisfactory yield (72%) from 55 and pinacol boronate 56, along with trace amounts of the double bond migrated side product 58 (57:58 = ca. 15:1). Demethylation of 57 to phenolic intermediate followed by the construction of the B ring generated tricyclic core 59. Subsequently, dihydroxylation of the doubled bond in the central six-membered ring using OsO_4_/NMO gave diol, which was then subjected to acetylation of the two hydroxy groups and hydrogenation of C5=C6 double bond to afford triacetate 60 as a single diastereoisomer. Base-promoted hydrolysis and concomitant oxidation under oxygen atmosphere gave vicinal diketone 61. Finally, the introduction of mono-bromo and di-bromo atoms achieved the total synthesis of (−)-hamigeran B (11) and (−)-4-bromohamigeran B (12), respectively.
The total synthesis of (−)-hamigeran B and (−)-4-bromohamigeran B by Han [51].
Total synthesis of (+)-randainin D
(+)-Randainin D (13) is a representative member of a class of structurally intriguing diterpenoids containing a trans-hydroazulenone core and a C9-butenolide moiety isolated from Callicarpa randaiensis [52]. It exhibits inhibition of elastase release and superoxide-anion generation. In 2024, Baudoin and co-workers presented an efficient route for the first total synthesis (+)-randainin D (13) by utilizing an early-staged Ru-catalyzed desymmetric enantioselective reduction as the key transformation [53].
As shown in Scheme 5 [53], based on the reported method for asymmetric transfer hydrogenation of commercially available cyclopentadione 62 [54], the authors adapted an efficient method for the desymmetric enantioselective reduction of 62 using commercially available (R,R)-Ts-DENEB (63) as the catalyst and formic acid as the hydrogen donor, securing the desired secondary alcohol product in excellent enantioselectivity (99% ee). Protection of the alcohol group with TBDPSCl gave silyl ether 64 in high yield (79% for 2 steps). Subsequently, successive four manipulations including dehydrogenation, Morita–Baylis–Hillman reaction, protection of the resultant primary alcohol, and hydrogenation afforded ketone 65. The LaCl_3_·LiCl-promoted addition of 65 with Grignard reagent followed by TES protection of the resulting secondary alcohol, regioselective deprotection of the TES group and in situ oxidation provided aldehyde 66. Next, 66 underwent the 1,2-addition of isopropenyllithium reagent (prepared from n-BuLi/tetraisopropenyltin (67) [55]) and DMP oxidation to afford ketone 68. A three-step transformation including RCM, Mukaiyama hydration, and esterification, 68 was converted to methyl oxalate 69. Irradiation of the reaction mixture 69 and lactone 70 at 456 nm with fac-Ir(ppy)3 as the photocatalyst furnished a mixture of isomeric olefins. Finally, DBU-promoted the isomeric olefins conjugation and removal of the two silyl ether completed the first total synthesis of (+)-randainin D (13).
The total synthesis of (+)-randainin D by Baudoin [53].
Total synthesis of (−)-hunterine A and (−)-aspidospermidine
(−)-Hunterine A (14) and (−)-aspidospermidine (15) are monoterpene indole alkaloids, which were isolated from Hunteria zeylanica and Apocynaceae plants, respectively [56–57]. Structurally, these two natural products both contain the fused polycyclic skeleton core bearing four consecutive stereocenters, two of which are all-carbon quaternary stereocenters. Such a skeletally extraordinary structure poses significant challenges for total synthesis. In 2024, Stoltz and co-workers finished the divergent enantioselective total synthesis of (−)-hunterine A and (−)-aspidospermidine by utilizing a Ru-catalyzed desymmetric enantioselective reduction of bicyclic 1,3-diketone 72 for rapidly accessing the 5/5 bicyclic core skeleton containing two desired stereocenters [58]. The application of this strategy accelerated the synthesis of these two complex alkaloids within a longest linear sequence of 16 and 14 steps, respectively.
As shown in Scheme 6 [58], the Cu-catalyzed 1,4-conjugated addition of 71 [59] with vinylMgBr followed by intramolecular Claisen condensation furnished bicyclic diketone 72 on a gram scale. Notably, for such a bicyclic substrate, it is quite challenging to achieve desymmetric enantioselective reduction because the catalyst has difficulty identifying its Re- and Si-faces. After screening several reduction strategies, the authors identified a set of Ru-catalyzed transfer hydrogenation conditions [60–61] utilizing 73 as the catalyst that could be used for the desymmetric enantioselective reduction of 72, affording the hydroxyketone 74 in 57% yield with 91% ee. Protection of the secondary alcohol in 74 followed by Beckmann rearrangement led to lactam 75. Oxidation state modifications and functional group transformations of 75 afforded ketone 76. Next, the 1,2-addition of 76 with vinyl iodine 77 and subsequent deprotection produced tertiary alcohol 78. Starting from this common intermediate, on the one hand, through successive manipulations by diazotization and in situ azide substitution, AgNO_3_-mediated aza-Cope/Mannich [62] reaction delivered ketone 79. Subsequently, a three-step operation including azide–alkene dipolar cycloaddition, irradiation of the resulting triazoline to aziridine 80 and in situ ring opening followed by deacetylation achieved the first total synthesis of (−)-hunterine A (14). On the other hand, aza-Cope/Mannich reaction of 78 produced imine intermediate 81. Reduction and hydrogenation of 81 furnished the total synthesis of (−)-aspidospermidine (15).
The total synthesis of (−)-hunterine A and (−)-aspidospermidine by Stoltz [58].
Total synthesis of terpenoid natural products through the desymmetric enantioselective reduction of six-membered cyclic 1,3-dicarbonyl compounds
Total synthesis of (+)-toxicodenane A
The skeletally new sesquiterpenoids, toxicodenanes A–C and E (see representative structure 16 in Scheme 7) were isolated from Toxicodendron vernicifluum in 2013 to 2015 [63–64], whose structures feature the all-carbon bicyclic skeleton and four to seven contiguous stereocenters, posing significant challenges to their synthesis. Additionally, there is a lack of corresponding pharmacological activity studies. In 2021, the group of Han completed the first enantioselective total synthesis of (+)-toxicodenane A (16) and determined its absolute configuration by employing an early-stage desymmetric enantioselective reduction of a 1,3-cyclohexanedione derivative as the key transformation [65]. The application of this strategy markedly accelerated the synthesis of the complex molecule within a longest linear sequence of 9 steps from the commercially available material.
The synthesis began with the commercially available 1,3-cyclohexanedione 82 (Scheme 7) [65–66]. Accordingly, the α-dialkylation of 82 gave diketone 83. Based on the conditions developed by the authors [67], namely, using P-stereogenic phosphinamide 84 as the catalyst and base 85 as the additive, the desymmetric enantioselective reduction of 83 proceeded smoothly to deliver the hydroxyketone 86 in 60% yield with 92% ee and 8.4:1 dr. A secondary hydroxy-directed Grignard reagent addition of 86 followed by selective protection, generated alcohol ester 87. After one recrystallization, the ee value of 87 could be increased to 99%. Subsequently, TMSOTf-promoted transacetalation and in situ Prins cyclization produced the oxa-bridged product 88. Finally, ozonolysis of 88 followed by Wittig reaction of the resultant ketone and subsequent reduction accomplished the total synthesis of (+)-toxicodenane A (16). The authors eventually confirmed that the absolute configuration of (+)-16 was 4S,5S,8R,11R based on the X-ray single-crystal analysis of its corresponding p-bromobenzoic ester (not shown).
The total synthesis of (+)-toxicodenane A by Han [65–66].
Total synthesis of (−)-conidiogenones B–F and (−)-12β-hydroxyconidiogenone C
Conidiogenones are unique diterpenoids which possess a highly congested 6/5/5/5-fused framework with four all-carbon quaternary centers isolated from Penicillium cylopium and exhibit various biological properties [68]. The intriguing structure and interesting biological properties have attracted continued synthetic attention [69–71]. In a 2023 report, the group of Lee and Han adopted an early-stage desymmetric enantioselective reduction of 1,3-cyclohexanedione derivative 89 as the key transformation [72]. Both (−)-conidiogenones B–F (17–21) and (−)-12β-hydroxyconidiogenone C (22) were synthesized in a divergent manner.
Their synthetic route began with the known terminal alkyne cyclohexanedione 89 [73]. As illustrated in Scheme 8 [72], the easily prepared substrate underwent the CBS reduction conditions and protection of the resulting secondary alcohol with TBSCl to afforded silyl ether ketone 90 in 46% yield (two steps, dr = 40:1, 92% ee) [8]. Horner–Wadsworth–Emmons (HWE) reaction of 90 followed by Cu-carbene migratory insertion [74] with ketone 91 and deprotection of the dithiane group delivered alleneketone 92. Sequential treatment of 92 with TsNHNH_2_ and NaH, the trimethylenemethane (TMM) diyl-mediated cycloaddition via intermediates 93 and 94 proceeded uneventfully to form the tetracyclic product 95. Next, reductive decyanation [75] and double bond migration of 95 produced common intermediate 96, which was elaborated to ketone 97 [69] via four functional group manipulations, thereby achieving the formal total synthesis of (−)-conidiogenone B (17). On the other hand, functionalization and derivatization were performed on the A- and D-rings of 96, respectively, delivering the methyl enol ether 98. Finally, allylic oxidation [76], α-methylation and reduction of 98 followed by hydrolysis accomplished the first total synthsis of (−)-conidiogenone F (21).
The formal total synthesis of (−)-conidiogeone B and total synthesis of (−)-conidiogeone F by Lee and Han [72].
After achieving the aforementioned success, the authors continued to utilize 96 as the common intermediate to synthesize other target natural products (Scheme 9) [72]. Firstly, the allylic oxidation and Luche reduction of 96 afforded primary alcohol 99, which was elaborated to (−)-conidiogenone D (19) via ten functional group manipulations. Secondly, the allylic oxidation of 96 with CrO_3_/3,5-DMP produced ketone 100. The 1,4-conjugate addition of 100 with MeI followed by Luche reduction provided the secondary alcohol 101. A-ring modifications in 101 completed the first total synthesis of (−)-conidiogenone E (20). One the other hand, a two-step transformation involving the 1,4-conjugate addition utilizing vinylMgBr and Luche reduction, 100 was converted to secondary alcohol 102 (dr = 2:1). Finally, functional group modifications of A- and D-rings furnished the total syntheses of (−)-conidiogenone C (18) and (−)-12β-hydroxyconidiogenone C (22), respectively.
The total syntheses of four conidiogenones natural products by Lee and Han [72].
Total syntheses of (−)-platensilin, (−)-platencin, and (−)-platensimycin
(−)-Platensilin (23), (−)-platencin (24), and (−)-platensimycin (25) are structurally unique meroterpenoids containing rigid bicycle [3.2.1] or [2.2.2]-octane cage core. They present promising drug leads for both antidiabetic and antibacterial therapies. The intriguing structural features and potential bioactivities have stimulated tremendous synthetic efforts [77–81]. In 2024, the group of Lou and Xu presented a unified and efficient route for the syntheses of (−)-platensilin, (−)-platencin, and (−)-platensimycin by utilizing a desymmetric enantioselective reduction of 1,3-cyclohexanedione derivative strategy [82].
The synthesis of (−)-platensilin (23) is shown in Scheme 10 [82]. Brewer′s yeast (YSC-2)-promoted desymmetric enantioselective reduction of 1,3-cyclohexanedione derivative 103 proceeded smoothly to produce hydroxyketone 104 in excellent diastereoselectivity and enantioselectivity [9,11]. Ozonolysis of the double bond in 104 followed by Purdie methylation with Ag_2_O/MeI, base-mediated vinyl triflation, and Pd-catalyzed Suzuki–Miyaura cross coupling with pinacol boronate 105 delivered diene 106. Next, The Et_2_AlCl-catalyzed intermolecular Diels–Alder reaction of 106 with methacrolein 107 afforded the common intermediate 108 in high yield. Sequential Grignard reagent addition and acid-promoted ethoxy elimination provided the separable planar diene 109 (dr = 1:1), which underwent a Mn-catalyzed HAT hydrogenation to give (15R)-110 and (15S)-110 in 65% and 54% yield, respectively. Subsequently, ten functional group manipulations of the diastereomeric mixture 110 produced ketoester 111. Finally, the introduction of conjugated double bond in 111 followed by hydrolysis of the methyl ester to carboxylic acid and DCC-mediated condensation with 112 accomplished the total synthesis of (−)-platensilin (23).
The total synthesis of (−)-platensilin by Lou and Xu [82].
Based on the aforementioned successful work, the authors focused on the synthesis of (−)-platencin (24) and (−)-platensimycin (25) (Scheme 11) [82]. Accordingly, Wittig reaction of 108 followed by 1,4-elimination and intramolecular Diels–Alder reaction generated tricyclo[3.2.1.0^2,7^]-octene 113. A two-step transformation including HAT hydrogenation and acetal C–H oxidation with RuCl_3_/NaIO_4_, 113 was converted into ketoester 114. The TFA-mediated C13–C15 bond cleavage of 114 proceed smoothly to give ring-opening products, which underwent dehydration with Martin′s sulfurane to afford the known intermediate 115 [83] and terminal alkene 116 (C12–C13 bond-cleaved byproduct). Thus, the formal total synthesis of (−)-platencin (24) was achieved. On the other hand, epoxidation of 113 followed by acid-mediated regioselective ring-opening and in situ allylic substitution with MeOH produced [3.2.1] bridged ring product 117, which was transformed into ketone 118 via nine functional group manipulations. Finally, by employing the same reaction procedures as those utilized in the total synthesis of (−)-platensilin (23), the authors accomplished the total synthesis of (−)-platensimycin (25).
The total synthesis of (−)-platencin and (−)-platensimycin by Lou and Xu [82].
Total synthesis of (+)-isochamaecydin and (+)-chamaecydin
(+)-Isochamaecydin (26) and (+)-chamaecydin (27) are represented members of the cryptoquinonemethides isolated from the seed of chamaecyparis obtusa Endl. genus [84]. This two compounds possess the unprecedented spiroannulated 6/6/6/5/5/3 (A/B/C/D/E/F) hexacarbocyclic motif and exhibit significant antifeedant activity against the pest insect Spodoptera litura [85]. In a very recent report, Han and co-workers accomplished the first catalytic asymmetric total syntheses of 26 and 27 via a modular and convergent strategy [86]. The key to their successful synthesis depended on the method application of a desymmetric enantioselective reduction of a 1,3-cyclohexanedione derivative as the key transformation.
Their synthesis began with the known 1,3-cyclohexanedione 119 (Scheme 12) [86]. Initially, for the construction of the crucial B ring bearing an all-carbon quaternary stereocenter, the authors employed a method they had previously reported [35,67]. Namely, the P-stereogenic phosphinamide (84, see Scheme 7)-catalyzed desymmetric enantioselective reduction of 119. Hydroxyketone 120 could be acquired in 60% yield with 93% ee. Subsequently, the Grignard addition of the ketone in 120 provided diene intermediate, which was converted to bicyclic secondary alcohol 121 via RCM reaction/hydrogenation and dehydration/hydrogenation. Oxidation states adjustment and functional group transformations of 121 generated bromodiene 122. Next, the metal–halogen exchange/intermolecular addition of 122 with aldehyde (+)-123 and in situ PtCl_2_-promoted hydrolysis and hydration gave tricyclic product 124. The BnMe_3_NOH-mediated intramolecular Michael/aldol cascade reaction of 124 constructed the C/D rings, followed by dehydration to afford pentacyclic product 125. Finally, a five-step operation including the 1,2-addition of 125 with iPrLi, PCC oxidation of the resulting tertiary alcohol, oxidative aromatization and in situ neutralization/oxidation, acid-mediated 1,6-addition with H_2_O, and stereoselective cyclopropanation accomplished the first total synthesis of (+)-isochamaecydin (26).
The total synthesis of (+)-isochamaecydin and (+)-chamaecydin by Han [86].
On the other hand, starting from 122 and (−)-123, the authors adopted the same procedures as for the synthesis of 125 to obtain the pentacyclic product 126, which underwent the same functional group transformations for the synthesis of (+)-26 to complete the first total synthesis of (+)-chamaecydin (27). The successful collective total synthesis of the two structurally complex natural products demonstrated the high efficiency of the modular synthetic strategy based on the desymmetric enantioselective reduction of cyclic 1,3-dicarbonyl compounds.
Summary and Outlook
In this review, we summarized the application of desymmetric enantioselective reduction of cyclic 1,3-dicarbonyl compounds in the total synthesis of a series of terpenoid and alkaloid natural products reported between 2016 and 2025. It was evident that the application frequency of this strategy in the synthesis of terpenoid and alkaloid natural products has increased significantly over these ten years compared to the period before 2016. Notably, the substrates of desymmetric enantioselective reduction were no longer limited to monocyclic 1,3-dicarbonyl compounds but could also be extended to bicyclic dicarbonyl compounds. The diversification of substrates enabled this strategy to provide a novel alternative for the synthetic design of structurally more complex natural products. More importantly, from these previous works, it could be observed that the early-stage application of desymmetric enantioselective reduction could efficiently provide access to enantiomerically enriched intermediates with multiple stereocenters containing an all-carbon quaternary chiral center. Taking aforementioned advantages of this strategy, the synthetic efficiency was significantly improved.
Despite remarkable progress has been made, there is still a long way to go before the strategy achieves maturity for applications in natural products. Specifically, challenges remain in improving the enantioselectivity and diastereoselectivity of the desymmetric enantioselective reduction of some complex substrates, as well as suppressing the double reduction by-products and developing new types of catalysts. Moreover, the application of desymmetric enantioselective reduction of chain dicarbonyl compounds in natural products synthesis remains largely undeveloped although several desymmetric enantioselective reduction studies utilizing malonate ester as substrates have been achieved [87–89]. Nevertheless, we believed on the basis of these pioneering works of the development of new reagents and methodologies for desymmetric enantioselective reduction based on the novel dicarbonyl substrates, as well as their applications in the synthesis of various complex natural products will become a focal point in the field of organic synthetic chemistry.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sun H-D Huang S-X Han Q-B Nat Prod Rep 20062367369810.1039/b 604174 d 17003905 · doi ↗ · pubmed ↗
- 2Liu M Wang W-G Sun H-D Pu J-X Nat Prod Rep 2017341090114010.1039/c 7np 00027 h 28758169 · doi ↗ · pubmed ↗
- 3O'Connor S E Maresh J J Nat Prod Rep 20062353254710.1039/b 512615 k 16874388 · doi ↗ · pubmed ↗
- 4Aoki S Watanabe Y Tanabe D Setiawan A Arai M Kobayashi M Tetrahedron Lett 2007484485448810.1016/j.tetlet.2007.05.003 · doi ↗
- 5Brooks D W Mazdiyasni H Chakrabarti S Tetrahedron Lett 1984251241124410.1016/s 0040-4039(01)80123-4 · doi ↗
- 6Shimizu M Yamada S Fujita Y Kobayashi F Tetrahedron: Asymmetry 2000113883388610.1016/s 0957-4166(00)00378-5 · doi ↗
- 7Hashiguchi S Fujii A Takehara J Ikariya T Noyori R J Am Chem Soc 19951177562756310.1021/ja 00133 a 037 · doi ↗
- 8Yeung Y-Y Chein R-J Corey E J J Am Chem Soc 2007129103461034710.1021/ja 074243417672468 · doi ↗ · pubmed ↗