Metatranscriptomic Analysis Uncovers RNA Virus Diversity in Ticks From the China–Russia–North Korea Border Region
Zhe Liu, Shengwei Ji, Jinqi Wang, Yuan Li, Eloiza May Galon, Shanshan Wang, Jixu Li, Xu Gao, Longzheng Yu, Yang Wang, Jianchen Song, Qichao Cui, Chenghui Li, Zhiqiang Xu, Shujiang Xue

TL;DR
This study used metatranscriptomics to discover new viruses in ticks from a border region, revealing potential zoonotic threats and ecological patterns.
Contribution
The study identifies novel tick-borne viruses and their ecological associations in a geopolitically significant border region.
Findings
Metatranscriptomic analysis identified 10 viral families and 22 viral species in five tick species.
Five viruses were confirmed human pathogens, and nine showed potential zoonotic risks.
Virome composition varied significantly across sampling sites, linked to ecological-geographical factors.
Abstract
Ticks serve as critical viral vectors, and border regions, acting as convergence zones of complex ecosystems, provide diverse habitats for ticks and their hosts, thereby underscoring the need to investigate the tick-borne virome composition in such areas. In this study, metatranscriptomic analysis of five tick species, namely Haemaphysalis longicornis, Haemaphysalis concinna, Haemaphysalis japonica, Ixodes persulactus, and Dermacentor silvarum, collected from the China–Russia–North Korea border region identified 10 viral families and 22 viral species. Among these, five were confirmed human pathogens, while nine exhibited potential zoonotic risks. Moreover, significant variations in virome composition across sampling sites revealed associations between tick-borne viruses and ecological-geographical factors. These findings highlight the diversity and spatiotemporal distribution patterns…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
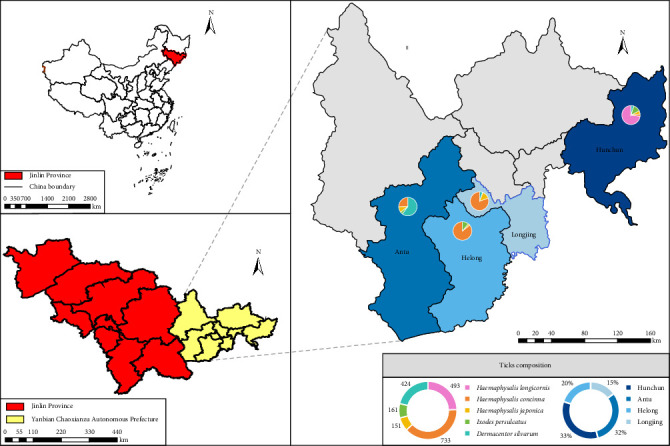 Figure 1
Figure 1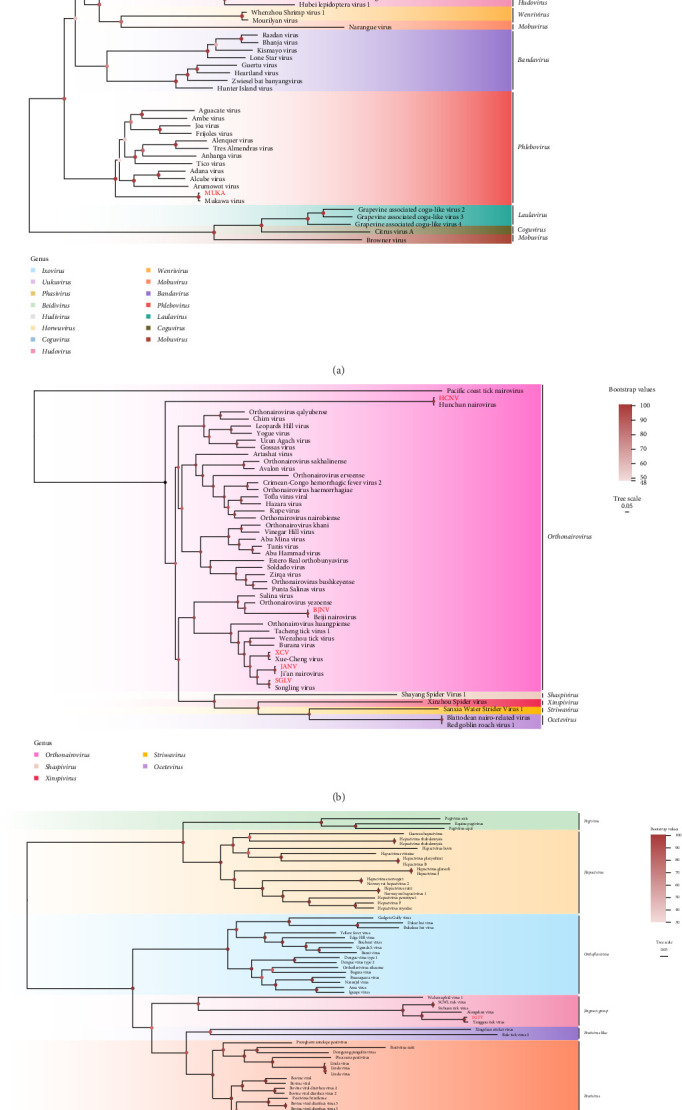 Figure 2
Figure 2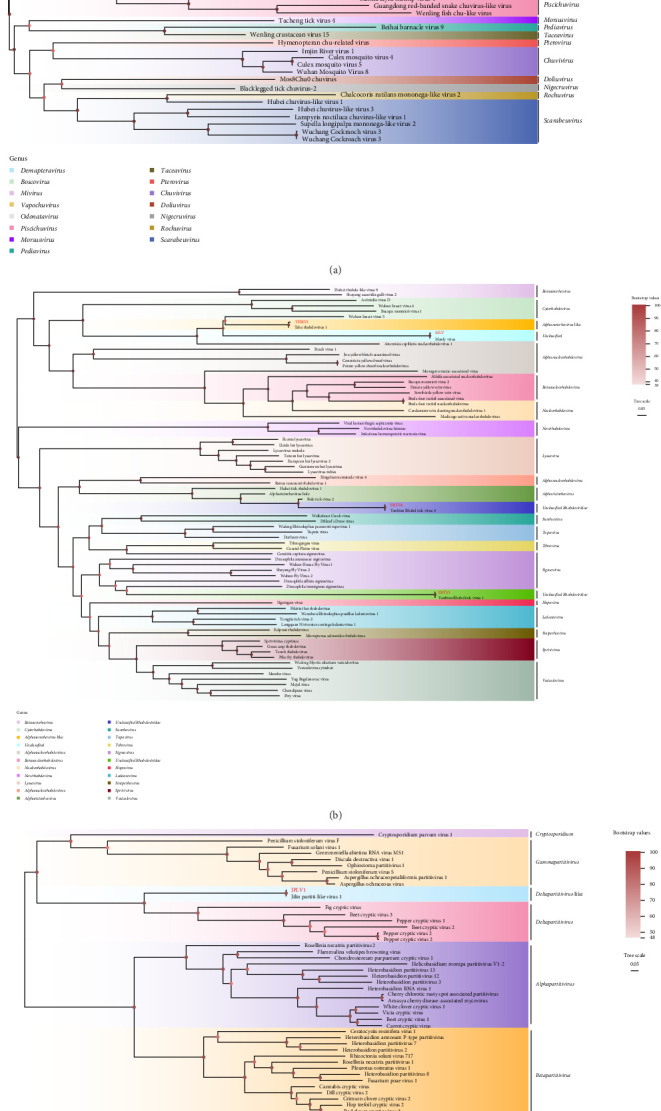 Figure 3
Figure 3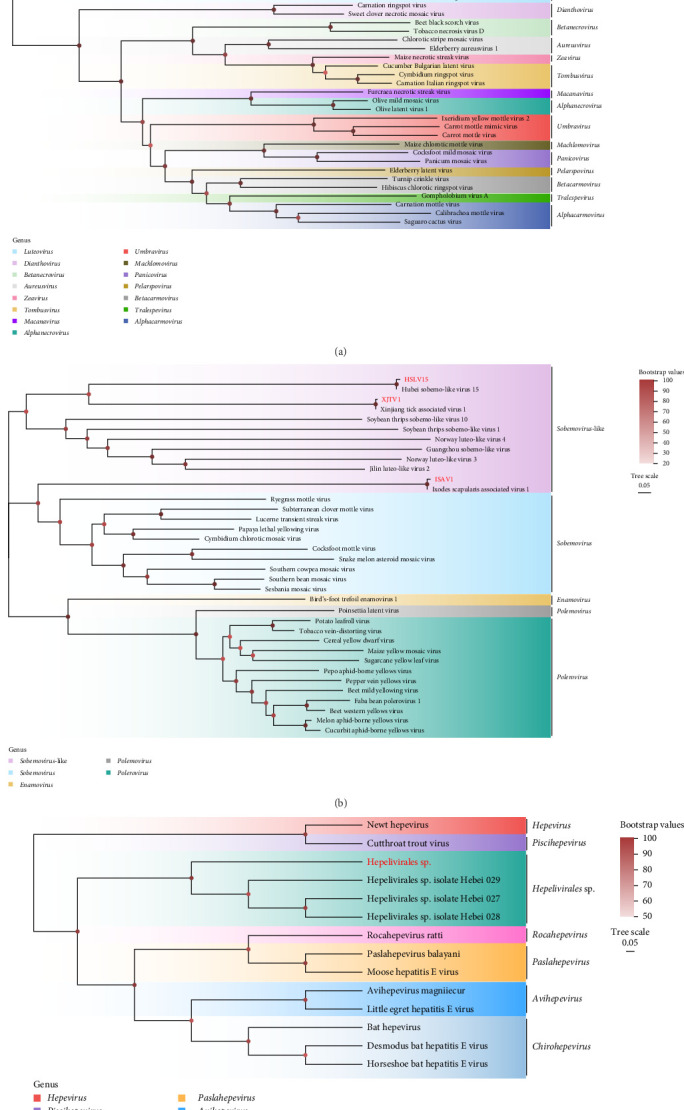 Figure 4
Figure 4- —National Natural Science Foundation of China
- —Natural Science Foundation of Jilin Province
- —Education Department of Jilin Province
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsViral Infections and Vectors · Plant Virus Research Studies · Vector-Borne Animal Diseases
1. Introduction
Ticks rank as the second most significant natural vectors of arboviral diseases, second only to mosquitoes, with over 800 species identified globally. These arthropods are capable of transmitting more than 200 pathogens, including over 160 tick-borne viruses, posing substantial threats to public health [1]. Approximately 30% of tick-borne viruses are associated with severe human diseases, exemplified by Xue-Cheng virus (XCV) [2], Songling virus (SGLV) [3], Beiji nairovirus (BJNV) [4, 5], tick-borne encephalitis virus (TBEV) [6–9], Wetland virus (WELV) [10], Crimean-Congo hemorrhagic fever virus (CCHFV) [11, 12], and Alongshan virus (ALSV) [13–15]. In recent years, intensified research on tick-borne pathogens has led to the discovery of numerous novel agents, coinciding with rising incidence rates of tick-borne diseases across regions. This trend reinforces the existence of a vast reservoir of undiscovered, potentially pathogenic tick-borne agents awaiting characterization [16–19].
Next-generation sequencing (NGS) has been extensively employed for monitoring pathogens in humans, animals, and plants [1]. In China, metatranscriptomic analysis of serum samples from febrile patients in 2025 first identified XCV, a novel tick-borne orthonairovirus belonging to the Orthonairovirus genus within the Nairoviridae family, with subsequent detection in Haemaphysalis concinna and H. japonica [2]. SGLV, initially discovered in a patient in 2021 [20], has since been documented in Xinjiang gerbils [21] and diverse tick species across multiple Chinese regions [22–24]. Similarly, WELV, a novel pathogen linked to multiorgan dysfunction, was first isolated via NGS analysis and is predominantly distributed in Inner Mongolia, Jilin, Liaoning, and Heilongjiang provinces [25]. Metagenomic sequencing has emerged as a pivotal tool for dynamically surveilling known and emerging pathogens, elucidating viral evolution patterns, and providing critical insights into transmission mechanisms and public health risk assessments.
The China–Russia–North Korea border region, characterized by rich biodiversity and frequent cross-border trade activities [26], is highly sensitive to climate change, with an annual temperature increase rate exceeding the national average. This climatic shift has prolonged tick activity periods, significantly elevating the risk of emerging infectious diseases [27]. To date, multiple zoonotic viruses have been identified in this area, including WELV [10], Tamdy orthonairovirus (TDOV) [20], TBEV [24], severe fever with thrombocytopenia syndrome virus (SFTSV) [28], Nairobi sheep disease virus (NSDV) [28], Onega tick phlebovirus (OTPV) [29], and ALSV [22]. These findings highlight the need to strengthen surveillance of tick-borne diseases in this ecologically vulnerable zone. In this study, we conducted molecular epidemiological investigations on ticks collected from the region through metatranscriptomic analysis, aiming to delineate the geographical distribution of tick species and their associated viral prevalence. Our objectives were to delineate tick species distribution and determine the prevalence of tick-borne viruses, thereby providing critical data for assessing cross-border transmission risks and formulating targeted biosecurity measures to mitigate public health threats.
2. Materials and Methods
2.1. Sample Collection and Identification
From April to July 2023 and 2024, ticks were collected from four regions (Antu County, Hunchun City, Helong City, and Longjing City) within the China–Russia–North Korea border region using the woolen flannel cloth dragging method (Figure 1). Following field collection, specimens were temporarily stored on dry ice during transportation and subsequently transferred to −80°C ultra-low temperature freezers for long-term preservation in laboratory facilities. Collected tick specimens underwent preliminary morphological identification via stereomicroscopy using taxonomic identification keys [30], followed by molecular confirmation through sequencing of the mitochondrial cytochrome c oxidase subunit I (COI) gene [31, 32].
2.2. RNA Library Construction and Sequencing
The collected tick specimens were categorized into 287 pools based on collection sites, species, and sex (Supporting Information 1). Specimens underwent sequential decontamination through immersion in 75% ethanol followed by three washes with phosphate-buffered saline (PBS, pH 7.4) [33]. Each pool was homogenized in 500 μL Dulbecco's Modified Eagle's Medium (DMEM) supplemented with two 3 mm zirconium beads using the high-throughput tissue grinder (Jingxin, China) at 60 Hz for 10 min. Homogenates were centrifuged at 13,400 × g for 15 min at 4°C to pellet cellular debris. Supernatants were sequentially filtered through 0.45 and 0.22 μm Millex membrane filters (Millipore, USA) to eliminate residual particulates. Filtered lysates were aliquoted into two fractions: one for RNA library construction and the other for virome characterization.
The 287 filtrate samples were categorized into four libraries based on sampling locations. Free nucleic acids in the filtrate were enzymatically digested prior to total RNA extraction using the QIAamp Viral RNA Mini Kit (Qiagen, Germany). Total RNA was subsequently fragmented, and double-stranded cDNA was synthesized via random hexamer priming and dNTP incorporation, followed by adapter ligation, single-primer PCR amplification, and purification according to the manufacturer's protocol. Metatranscriptomic analysis was conducted by Shanghai Personalbio Technology Co., Ltd. utilizing the Illumina NovaSeq 6000 platform.
2.3. Bioinformatics Analysis
Raw sequencing data from each library were processed using fastp (v0.20.0) [34] to remove low-quality sequences, including short reads and adapter-contaminated fragments. Cleaned reads were then aligned to the host genome using minimap2 (v2.24) [35] to eliminate host-derived contamination. De novo assembly of the remaining high-quality reads was performed with MEGAHIT (v1.2.9) [36] under the meta-sensitive preset parameters, retaining contigs ≥300 bp in length. To recover fragmented sequences, unmapped reads from each sample were realigned to their corresponding contigs using minimap2 and subjected to iterative reassembly. Taxonomic annotation of contigs was conducted via Kaiju [37] and DIAMOND BLASTp [38] against the NCBI nr and RefSeq-Viral databases, with stringent thresholds set at a maximum of five mismatches and an E-value cutoff of 1 × 10^−5^ to minimize false-positive identifications [37, 39].
2.4. Viral Screening
To validate the Illumina sequencing results, primers were designed using Primer 6 (Premier Biosoft International, USA; primer sequences listed in Supporting Information 2). Viral RNA was extracted using the QIAamp Viral RNA Mini Kit (Qiagen, Germany), followed by cDNA synthesis with FastKing gDNA Dispelling RT SuperMix (Tiangen, China). PCR amplification was performed using PCR Master Mix (Tiangen, China), and all amplicons were resolved on 1.0% agarose gels. Positive amplification products were purified using the TIANgel Midi Purification Kit (Tiangen, China) and subsequently subjected to Sanger et al. [40] sequencing for verification.
2.5. Phylogenetic Analysis
To confirm the phylogenetic relationships of the viral strains identified in this study, complete and partial nucleotide sequences were subjected to BLASTn analysis against the NCBI database to retrieve homologous reference sequences. Multiple sequence alignment was performed using MAFFT v7.526 [41], followed by maximum-likelihood phylogenetic tree construction with IQ-TREE v2.4.0 [42] under 1000 ultrafast bootstrap replicates. Final tree visualization and annotation were implemented in FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/), with midpoint rooting applied in the absence of a predefined outgroup and nodes ordered by descending support values.
2.6. Statistical Analysis
The viral infection rates of ticks were calculated using PooledInfRate software, version 4.0 (a Microsoft Office Excel Add-In designed by Brad J. Biggerstaff to compute prevalence estimates from pooled samples, Centers for Disease Control and Prevention, Fort Collins, CO, USA, 2024). The prevalence of viruses in ticks was presented as the maximum likelihood estimate (MLE) per 100 ticks with a 95% confidence interval (95% CI). The data were analyzed using SPSS version 21.0.
3. Results
3.1. Tick Collection and Identification
A total of 1962 ticks were collected from the China–Russia–North Korea border region and classified into five species: H. longicornis (n = 493), H. concinna (n = 733), H. japonica (n = 151), I. persulcatus (n = 161), and D. silvarum (n = 424) (Figure 1 and Table 1). Sampling sites included Helong City (n = 416), Hunchun City (n = 676), Antu County (n = 578), and Longjing City (n = 292) (Supporting Information 1). Analysis revealed that H. concinna, H. japonica, and I. persulcatus were distributed across all four sampling sites, whereas H. longicornis was exclusively detected in Hunchun City.
3.2. Detection and Statistical Analysis of RNA Viruses
Four RNA virome libraries were constructed and sequenced, generating 90 GB of raw data. Following quality filtering, 1,727,344 clean reads were retained. De novo assembly yielded 4526 viral contigs, with viral reads representing 0.06%–0.75% of non-rRNA reads per library. Viral sequences were annotated into nine viral families and one unclassified family, encompassing 22 viral species (Table 2), based on BLASTn analysis (E-value ≤1 × 10^−5^). Notably, five RNA virus families with zoonotic potential were identified: Flaviviridae, Nairoviridae, Phenuiviridae, Chuviridae, and Rhabdoviridae. Specific viruses included XCV, SGLV, Dabieshan tick virus (DBTV), Mukawa virus (MKWV), BJNV, Manly virus (MLV), Sara tick phlebovirus (STPV), Hunchun nairovirus (HCNV), Ji'an nairovirus (JANV), Yanbian Rhabd tick virus 1 (YBRV1), Yanbian Rhabd tick virus 4 (YBRV4), Tahe rhabdovirus 1 (THRV1), Lesnoe mivirus (LMV), and Yanggou tick virus (YGTV). Analysis of viral read abundance revealed that the genus Orthonairovirus within the family Nairoviridae exhibited the highest abundance, primarily represented by SGLV. First identified in 2021, SGLV is transmissible via ticks to diverse animals and humans, inducing febrile illnesses and headaches, thereby posing a significant public health threat [20]. The second most abundant reads were attributed to YGTV, a member of the Jingmenvirus group within the family Flaviviridae. To validate the virome results, we performed PCR assays for dual verification of the aforementioned viruses and systematically analyzed their geographic and tick-species distribution patterns (primer sequences are detailed in Supporting Information 2). All amplified sequences have been deposited in the NCBI database (GenBank accession numbers are available in Table 3 and Supporting Information 3).
Viral species and prevalence demonstrated marked geographic heterogeneity and tick-species specificity across four sampled regions. Hunchun exhibited the highest viral diversity with 14 species detected, while Helong showed the lowest, containing only three species (Supporting Information 4). Notably, only SGLV and LMV were detected in all four regions. LMV reached its highest prevalence in Helong (3.40%, 13/58), whereas SGLV peaked in Longjing (5.80%, 14/40). Additionally, a novel human-associated virus, XCV, was identified in Hunchun with a prevalence of 0.30% (2/107). Other viruses with zoonotic potential detected in this region included DBTV (5.77%, 31/107), HCNV (0.60%, 4/107), JANV (0.29%, 2/107), and STPV (0.15%, 1/107), with DBTV being the most prevalent. Antu harbored the highest variety of potentially human-infective viruses, encompassing SGLV (1.23%, 7/82), STPV (0.35%, 2/82), YGTV (0.35%, 2/82), THRV1 (0.35%, 2/82), YBRV4 (0.35%, 2/82), YBRV1 (0.17%, 1/82), and MLV (0.35%, 2/82), where SGLV showed the highest prevalence and YBRV1 the lowest. Helong and Longjing each yielded one human-associated virus: BJNV (0.24%, 1/58) and MKWV (0.34%, 1/40), respectively (Table 4; Supporting Information 5).
Several viruses demonstrated distinct geographic restrictions: BJNV was exclusively detected in Helong; DBTV, XCV, HCNV, JANV, CLTV3, HNTV, Hepelivirales sp., and ISAV1 were found only in Hunchun; YGTV, THRV1, YBRV1, YBRV4, and MLV were unique to Antu; while MKWV and NXLV were solely present in Longjing.
Analysis at the tick species level revealed that I. persulcatus carried the highest viral diversity, with eight viruses detected. In Hunchun, I. persulcatus harbored six viruses: SGLV (1.19%, 1/15), HCNV (5.00%, 4/15), STPV (1.23%, 1/15), JANV (1.14%, 1/15), JLPV1 (1.14%, 1/15), and ISAV1 (1.14%, 1/15), with HCNV exhibiting the highest prevalence. I. persulcatus from Antu carried three human-associated viruses—SGLV, YBRV1, and STPV—among which STPV showed the highest prevalence (9.11%, 2/9). Only BJNV was detected in I. persulcatus from Helong, at a prevalence of 2.19% (1/11), whereas no viruses were identified in I. persulcatus from Longjing. Among all viruses, the highest prevalence was observed for LMV in H. japonica from Helong (43.82%, 4/6), and the lowest for both MLV and THRV1 in D. silvarum from Antu (0.27%, 1/44 each) (Supporting Information 6).
3.3. RNA Viruses Diversity and Evolution
3.3.1. Phenuiviridae
The family Phenuiviridae, classified within the order Bunyavirales, comprises single-stranded negative-sense RNA viruses and encompasses several genera, including Phlebovirus, Bandavirus, Uukuvirus, Ixovirus, and Hudivirus [43–45]. In this study, three Phenuiviridae were identified: DBTV (520 bp), MKWV (895 bp), and STPV (443 bp). Among these, DBTV, phylogenetically assigned to the genus Uukuvirus, was exclusively detected in H. longicornis from the Hunchun region, exhibiting nucleotide sequence similarities of 98.80%–99.31% with other strains isolated from the same tick species. Similarly, MKWV was uniquely identified in D. silvarum from Longjing, sharing nucleotide identities of 98.18%–98.72% with other strains. In contrast, STPV, classified under the Ixovirus genus, was detected in I. persulcatus across both the Hunchun and Antu regions, showing 92.75%–99.78% nucleotide identity with other strains (Figure 2A and Table 2).
3.3.2. Nairoviridae
The genus Orthonairovirus within the family Nairoviridae comprises at least seven viral clusters encompassing 15 species, many of which pose significant threats to human and animal health [5, 46, 47]. These viruses are primarily transmitted by ticks and possess segmented single-stranded RNA genomes. In this study, five Orthonairovirus species were identified in tick specimens: SGLV (500 bp), BJNV (498 bp), HCNV (457 bp), JANV (1375 bp), and XCV (766 bp). SGLV was detected in H. concinna from Helong, Longjing, and Antu, as well as in I. persulcatus from Antu and H. longicornis from Hunchun, exhibiting nucleotide identities of 96.92%–99.45% with other strains. HCNV and BJNV were exclusively identified in I. persulcatus from Hunchun and Helong, respectively, while JANV was detected in both H. concinna and I. persulcatus from Hunchun, with nucleotide identities of 98.99%–99.82%, 99.18%, and 99.18%–99.52%, respectively. Additionally, XCV, a recently discovered zoonotic virus, was identified in H. japonica and H. concinna from Hunchun, sharing 96.88%–99.03% nucleotide identity with othoer strains isolated from H. concinna (Figure 2B and Table 2).
3.3.3. Flaviviridae
The family Flaviviridae comprises single-stranded positive-sense RNA viruses and encompasses four primary genera along with unclassified groups, including Orthoflavivirus, Hepacivirus, Pestivirus, Pestivirus-like, Pegivirus, and the Jingmenvirus group [48]. In recent years, Flaviviruses have been increasingly implicated in global outbreaks, posing significant threats to both human and animal health [13]. In this study, a segmented Flavivirus, YGTV (300 bp), was identified in D. silvarum from the Antu region. YGTV exhibited nucleotide sequence similarities of 96.72%–99.67% with other YGTV strains (Figure 2C and Table 2).
3.3.4. Chuviridae
The virus was initially discovered in arthropods from Xinjiang, China, and includes members such as Mivirus, Demapteravirus, Boscovirus, Morsusvirus, and Scarapeuvirus [49]. In this study, LMV (1281 bp), classified under the genus Mivirus within the family Chuviridae, was detected in H. concinna, H. japonica, and D. silvarum from Longjing, and presented 98.75%–100% nucleotide identity with other strains (Figure 3A and Table 2).
3.3.5. Rhabdoviridae
Members of the family Rhabdoviridae exhibit an exceptionally broad host range, infecting mammals, arthropods, fish, and plants [50–52]. These viruses possess single-stranded negative-sense RNA genomes characterized by diverse structural features [33]. In this study, three Rhabdoviruses were detected in ticks from the Antu region: THRV1 (239 bp), Alphanemrhavirus-like group, and two unclassified Rhabdovirids, YBRV4 (579 bp), YBRV1 (822 bp) and MLV (764 bp). The nucleotide identities of these strains were 99.37%–99.61%, 99.04%–99.54%, 99.70%–99.74%, and 99.68%, respectively (Figure 3B and Table 2).
3.3.6. Partitiviridae
The family Partitiviridae, encompassing double-stranded RNA viruses, includes genera such as Deltapartitivirus-like, Cryspovirus, Alphapartitivirus, Betapartitivirus, Deltapartitivirus, and Gammapartitivirus [53]. In this study, Jilin partiti-like virus 1 (JLPV1) (345 bp), Deltapartitivirus-like, was detected in I. persulcatus from both Hunchun and Antu. JLPV1 shared nucleotide identities of 99.68% with other strains (Figure 3C and Table 2).
3.3.7. Tombusviridae
The family Tombusviridae, known to primarily infect plants and induce symptoms such as leaf mottling, chlorosis, and localized necrosis, has been reported across multiple European countries and North America [54]. In this study, two viruses belonging to Luteovirus within this family―CLTV3 (1436 bp) and NXLV (1439 bp)―were identified in ticks. NXLV was detected in H. japonica from Longjing, exhibiting nucleotide sequence identities of 97.78%–98.09% with other strains isolated from H. japonica. CLTV3, identified in H. longicornis from Hunchun, shared nucleotide identities of 96.19%–99.31% with previously discovered strains detected in H. longicornis (Figure 4A and Table 2).
3.3.8. Solemoviridae
Viruses within the family Solemoviridae are predominantly distributed in tropical and subtropical regions and rank among the most devastating plant pathogens globally [55, 56]. Researchers identified 22 putative novel Solemoviridae in 2025 [57]. In this study, three viruses belonging to the Sobemo-like family―Xinjiang tick associated virus 1 (XTAV1) (754 bp), Hubei sobemo-like virus 15 (HSLV15) (534 bp), and ISAV1 (324 bp)―were detected. XTAV1 was exclusively identified in D. silvarum from the Antu region, exhibiting nucleotide sequence identities of 98.59%–99.42% with other strains isolated from the same tick species. HSLV15 was detected in H. concinna, H. japonica, and D. silvarum ticks from Longjing, as well as in H. japonica and H. longicornis from Hunchun, with shared nucleotide identities ranging from 97.07%–98.70% with other strains. ISAV1, detected in I. persulcatus from Hunchun, shared 98.79% nucleotide identity with other strains (Figure 4B and Table 2).
3.3.9. Hepeviridae
The family Hepeviridae comprises multiple genera, including Hepevirus, Hepelivirales sp., Piscihepevirus, Avihepevirus, Chirohepevirus, and Orthohepevirus [58]. In this study, contigs classified under Hepelivirales sp. (879 bp) were identified in H. longicornis from Hunchun. Hepelivirales sp. shared 96.14%–98.73% nucleotide identity with previously discovered strains detected in H. longicornis.
4. Discussion
Recent advances in metatranscriptomic analysis have facilitated the discovery of an increasing number of novel tick-borne viruses, significantly enriching the diversity of tick-borne viral species [44, 59–62]. In this study, metatranscriptomic analysis of ticks from the China–Russia–North Korea border region provided the first comprehensive characterization of viral composition in this ecologically sensitive area. In the five tick species analyzed, we identified 22 viral species spanning 10 families: Flaviviridae, Nairoviridae, Phenuiviridae, Chuviridae, Rhabdoviridae, Tombusviridae, Partitiviridae, Solemoviridae, Hepeviridae, and unclassified. Among these, five species are confirmed human pathogens, while nine exhibit potential zoonotic risks.
Previous metatranscriptomic analysis of I. persulcatus in the China–Russia–North Korea border region identified YGTV, STPV, MKWV, and BJNV, all of which were reconfirmed in this study [33]. Notably, XCV, a novel tick-borne Orthonairovirus initially discovered in a febrile patient, in H. concinna, and H. japonica from Mudanjiang City, Heilongjiang Province in 2025 [2], was also detected in H. japonica and H. concinna from Hunchun. This finding corroborates prior reports and suggests potential geographical expansion beyond the originally identified Mudanjiang focus, indicating broader regional dissemination. DBTV, currently widespread in Japan [63] and Chinese provinces such as Shandong and Hubei [18, 64, 65], has been classified within the Orthohantavirus dabieshanense species and is closely related to Hantaan virus (HTNV). It has been stated that DBTV can cause hemorrhagic fever with renal syndrome (HFRS) in humans [66]. Additionally, seropositivity for DBTV antibodies was detected in Shandong sheep sera [67]. Han et al. [66] postulated a coastal distribution pattern for DBTV, with prevalence correlating strongly with habitats suitable for its primary vector H. longicornis, and migratory bird activity as a key dispersal drivers [68]. Bai et al. [28] reported high DBTV prevalence in Liaoning border ticks, consistent with our findings of elevated positivity rates in H. longicornis from Hunchun. The broad distribution of DBTV underscores the urgent need to elucidate its pathogenesis and genetic traits while addressing risks of cross-border spread via wildlife migration or trade activities. In 2017, Li et al. [69] investigated SFTSV in ticks from the Yanbian region, reporting a minimum infection rate of 1.81%. Notably, no SFTSV-positive ticks have been reported in subsequent studies conducted post-2017 in this region, a finding corroborated in the current study. SGLV was detected across all four sampling sites, with H. concinna identified as the predominant tick species. Higher SGLV positivity rates were observed in Helong and Longjing, whereas lower prevalence was recorded in neighboring Hunchun and Antu, likely attributable to localized ecological heterogeneity among the regions. In 2023, Li et al. [23] identified a novel Orthonairovirus, Antu virus, in D. silvarum from the China–North Korea border region of Jilin Province. Phylogenetic analysis revealed its close relationship to SGLV, suggesting potential zoonotic capacity. However, Antu virus was not detected in D. silvarum from Antu in the current study, possibly due to temporal variations in tick sampling or degradation of low-titer viral RNA under noncontrolled conditions [70]. BJNV, classified under the genus Orthonairovirus within the family Nairoviridae, lacks the medium (M) genomic segment encoding viral glycoproteins [10]. First identified in I. persulcatus in 2014 [10, 22, 24], BJNV was later reported in China–North Korea border regions by Wang et al. [29] in 2017. To date, BJNV has only been documented in northeastern China. Our findings confirm its persistence in I. persulcatus from Helong in 2025, indicating the establishment of stable natural foci in this region. The maintenance of BJNV may involve vertical transmission in ticks or sustained multihost cycles among wildlife reservoirs, underscoring the necessity for continuous surveillance of tick-borne pathogens.
Furthermore, molecular epidemiological investigations of tick-borne viruses across four sampling sites in the China–Russia–North Korea border region revealed host-specific viral distributions. For instance, XTAV1 and STPV were exclusively detected in D. silvarum, while DBTV was restricted to H. longicornis, consistent with findings by Xu et al. [65]. This phenomenon may be attributed to the virus's reliance on amplifying hosts that have established a closed-loop transmission cycle exclusively with H. longicornis, while other tick species exhibit inefficient viral acquisition or transmission due to low host contact frequency. Additionally, transovarial transmission of DBTV in H. longicornis [64] could further explain its host-restricted distribution [71]. Notably, viral prevalence exhibited marked geographic heterogeneity: tick pools from Hunchun, Helong, Longjing, and Antu showed positivity rates of 11.33% (53/107), 6.13% (21/58), 14.45% (27/40), and 4.49% (23/82), respectively. Rhabdoviridae and Flaviviridae were uniquely identified in Antu, whereas Hepeviridae was confined to Hunchun. These patterns likely arise from multifactorial drivers, including animal migration, climatic variability, anthropogenic activities, and habitat modifications [72].
This study has several limitations. While we confirmed the presence of several human disease-associated viruses in the region, several previously documented tick-borne viruses—including Antu virus [23], Tacheng tick virus-1 (TcTV-1) [22], ALSV [29], and NSDV [28]—were undetected. This discrepancy may be attributed to multifactorial influences, including potential mismatches between the spatiotemporal sampling coverage and viral activity cycles, as well as genetic variations arising from viral mutation or evolutionary divergence. Thus, expanding the geographical scope of metatranscriptomic analyses and optimizing cross-seasonal sampling strategies will facilitate deeper elucidation of viral transmission dynamics. Furthermore, the absence of systematic environmental parameters (e.g., humidity and forest coverage) and human activity data from sampling sites constrains deeper exploration of viral dissemination patterns and evolutionary trajectories, which will constitute a priority for future investigations.
5. Conclusions
In conclusion, this study conducted metatranscriptomic analyses and prevalence surveys of tick-borne viruses in the China–Russia–North Korea border region, providing the first systematic assessment of the tick-borne virome in this area and revealing their potential public health risks. These findings not only enhance our capacity to predict the spatiotemporal distribution and transmission dynamics of these viruses but also lay a critical data foundation for cross-border joint prevention and control strategies, as well as risk assessment and early warning of emerging infectious diseases.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kong Y. Zhang G. Jiang L. Metatranscriptomics Reveals the Diversity of the Tick Virome in Northwest China Microbiology Spectrum 202210510.1128/spectrum.01115-22e 0111522 PMC 960266436214702 · doi ↗ · pubmed ↗
- 2Zhang M. Z. Bian C. Ye R. Z. Human Infection With a Novel Tickborne Orthonairovirus Species in China New England Journal of Medicine 2025392220020210.1056/NEJ Mc 241085339778175 · doi ↗ · pubmed ↗
- 3Gui Z. Ren Y. Guo Q. Development of a LAMP Assay for the Rapid Visual Detection of the Emerging Tick-Borne Songling Virus Parasites & Vectors 202417110.1186/s 13071-024-06552-7447 PMC 1152901639487524 · doi ↗ · pubmed ↗
- 4Ergunay K. Bourke B. P. Linton Y.-M. Bourret T. J. Exploring the Potential of Tick Transcriptomes for Virus Screening: A Data Reuse Approach for Tick-Borne Virus Surveillance P Lo S Neglected Tropical Diseases 202519310.1371/journal.pntd.0012907 e 0012907 PMC 1192220840048471 · doi ↗ · pubmed ↗
- 5Kishimoto M. Itakura Y. Tabata K. A Wide Distribution of Beiji Nairoviruses and Related Viruses in Ixodes Ticks in Japan Ticks and Tick-borne Diseases 202415610.1016/j.ttbdis.2024.10238010238038996644 · doi ↗ · pubmed ↗
- 6Chevalier N. MignéC. V. Mariteragi-Helle T. Seroprevalence of West Nile, Usutu and Tick-Borne Encephalitis Viruses in Equids From South-Western France in 2023 Veterinary Research 202556110.1186/s 13567-025-01508-w 91PMC 1202338540275349 · doi ↗ · pubmed ↗
- 7Hassall R. M. Holding M. Medlock J. M. Identifying Hotspots and Risk Factors for Tick-Borne Encephalitis Virus Emergence at Its Range Margins to Guide Interventions, Great Britain Eurosurveillance 2025301310.2807/1560-7917.ES.2025.30.13.2400441 PMC 1196996040183125 · doi ↗ · pubmed ↗
- 8Pulkkinen L. I. A. Butcher S. J. Anastasina M. Tick-Borne Encephalitis Virus: A Structural View Viruses 201810710.3390/v 100703502-s 2.0-85049520665350 PMC 607126729958443 · doi ↗ · pubmed ↗