Acute Myeloid Leukemia With RUNX1::RUNX1T1 Fusion Transformed From JAK2V617F-Mutated Polycythemia Vera: A Case Report
Hidenori Tsukiji, Seiko Miyazaki, Tadafumi Iino, Goichi Yoshimoto, Yasushi Kubota

TL;DR
A patient with polycythemia vera developed acute myeloid leukemia with a rare RUNX1::RUNX1T1 fusion gene.
Contribution
This is a rare case report of AML with RUNX1::RUNX1T1 fusion transforming from JAK2V617F-mutated polycythemia vera.
Findings
AML with RUNX1::RUNX1T1 fusion was diagnosed following a 22-year history of polycythemia vera.
The JAK2V617F mutation was detected at the time of transformation to AML.
Morphological and genetic findings confirmed the leukemic transformation from MPN.
Abstract
Leukemic transformation is an event that significantly affects the prognosis of patients with Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs). In acute myeloid leukemia (AML) transformed from MPNs, balanced chromosomal translocations, including t(8;21)(q22;q22.1), are extremely rare. A case of secondary AML with RUNX1::RUNX1T1 fusion that evolved from polycythemia vera (PV) is presented. An 87-year-old Japanese man was diagnosed with PV 22 years earlier and had been treated with hydroxyurea and aspirin. Twelve years earlier, a homozygous JAK2V617F mutation was identified. Regular blood tests conducted every four months showed leukocytosis (white blood cell count, 14.6 × 109/L), anemia (hemoglobin, 106 g/L), and thrombocytopenia (platelet count, 73 × 109/L). Of the white blood cells, 5.5% were blasts, and the pseudo-Pelger-Huët anomaly and degranulated neutrophils…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
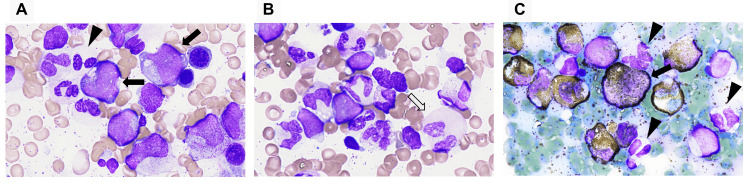 Figure 1
Figure 1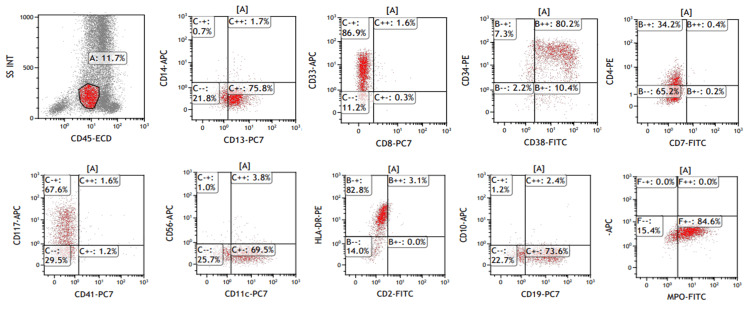 Figure 2
Figure 2 Figure 3
Figure 3| Parameters | Patient Values | Units | Reference Ranges |
| Complete blood count | |||
| WBC | 14.6 × 109 | /L | 3.3–8.6 × 109 |
| Blast | 5.5 | % | – |
| Myelocyte | 1.0 | % | – |
| Segmented neutrophils | 74.5 | % | 38.0–74.0 |
| Lymphocytes | 9.5 | % | 16.5–49.5 |
| Monocytes | 0.5 | % | 2.0–10.0 |
| Eosinophils | 8.00 | % | 0.0–8.5 |
| Basophils | 1.0 | % | 0.0–2.5 |
| RBC | 2.97 × 1012 | /L | 4.35–5.55× 1012 |
| Hemoglobin | 106 | g/L | 137–168 |
| Hemacrit | 31.9 | % | 40.7–50.1 |
| MCV | 107.4 | fL | 83.6–98.2 |
| MCH | 35.7 | pg | 27.5–33.2 |
| MCHC | 33.2 | g/dL | 31.7–35.3 |
| Platelet Count | 73 × 109 | /L | 158–348× 109 |
| Biochemistry | |||
| Total protein | 7.0 | g/dL | 6.6–8.1 |
| Albumin | 3.9 | g/dL | 4.1–5.1 |
| Total Bilirubin | 0.4 | mg/dL | 0.4–1.5 |
| AST | 23 | U/L | 13–30 |
| ALT | 28 | U/L | 10–42 |
| LDH | 472 | U/L | 124–222 |
| Creatine kinase | 67 | U/L | 59–248 |
| Urea nitrogen | 20 | mg/dL | 8.0–20.0 |
| Creatinine | 1.28 | mg/dL | 0.65–1.07 |
| Uric Acid | 7.5 | mg/dL | 3.7–7.8 |
| Sodium | 136 | mmol/L | 138–145 |
| Potassium | 5.2 | mmol/L | 3.6–4.8 |
| Clorine | 106 | mmol/L | 101–108 |
| Calcium | 8.8 | mg/dL | 8.8–10.1 |
| Glucose | 119 | mg/dL | 73–109 |
| CRP | 0.13 | mg/dL | 0.00–0.14 |
| Bone marrow exam | |||
| NCC | 30.0 × 104 | /μL | 10.0–25.0 × 104 |
| Megakaryocytes | 7 | /μL | 50–150 |
| M/E ratio | 21.0 | 1.1–3.5 | |
| Neutrophilic series | 87.6 | % | 34.7–78.8 |
| Myeloblast | 18.8 | % | 0.1–0.7 |
| Promyelocytes | 4.8 | % | 1.9–4.7 |
| Myelocytes | 4.8 | % | 8.5–16.9 |
| Metamyelocytes | 4.4 | % | 7.1–24.7 |
| Band neutrophils | 4.0 | % | 9.4–15.4 |
| Segmented neutrophils | 28.8 | % | 3.8–11.0 |
| Eosinophils | 13.6 | % | 1.1–5.2 |
| Basophils | 8.4 | % | < 0.1 |
| Erythrocytic series | 4.4 | % | 15.0–36.2 |
| Pronormoblasts | 0.0 | % | 0.1–1.1 |
| Basophilic erythroblasts | 2.4 | % | 0.4–2.4 |
| Polychromatophilic erythroblasts | 1.4 | % | 13.1–30.1 |
| Orthochromatic erythroblasts | 0.6 | % | 0.3–3.7 |
| Monocytes | 4.8 | % | 0.0–0.6 |
| Lymphocytes | 3.2 | % | 8.6–23.8 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMyeloproliferative Neoplasms: Diagnosis and Treatment · Acute Myeloid Leukemia Research · Chronic Myeloid Leukemia Treatments
Introduction
Polycythemia vera (PV) is a type of myeloproliferative neoplasm (MPN) that is Philadelphia (Ph) chromosome-negative, along with essential thrombocythemia (ET) and primary myelofibrosis (PMF). Over 95% of cases have a mutation in the JAK2 gene. PV is characterized by persistent activation of the JAK-STAT signaling pathway, leading to erythrocytosis as the primary feature, accompanied by leukocytosis and thrombocytosis, with an annual incidence of 0.5-4.0 cases per 100,000 persons. The disease typically progresses slowly, but during its course, splenomegaly or thrombosis may develop, and in some cases, it may transform to myelofibrosis (MF) or acute myeloid leukemia (AML) [1,2].
In contrast, AML with the RUNX1::RUNX1T1 fusion gene has a relatively good prognosis and is frequently observed in de novo AML [3], but transformation from MPN to AML with the RUNX1::RUNX1T1 fusion is extremely rare [4]. Herein, we present an exceptionally rare case of AML with RUNX1::RUNX1T1 fusion that transformed from JAK2V617F-mutated PV after a 22-year course, and we discuss the diagnostic clues and clinical implications.
Case presentation
An 87-year-old Japanese man was diagnosed with PV according to the Polycythemia Vera Study Group criteria in 2002 [5] and started on hydroxyurea therapy. His previous medical history was unremarkable. Phlebotomy was performed several times immediately after PV diagnosis. Subsequently, hematocrit levels remained between 45% and 50% through oral administration of hydroxyurea. He experienced transient ischemic attacks in 2007 and 2011, but no recurrences occurred. In 2012, he was found to be positive for the JAK2V617F gene mutation (allele burden was over 80%, homozygous), and aspirin therapy was started. At that time, bone marrow examination showed hyperplasia, with cells at various stages of maturation observed in all three hematopoietic lineages. No increase in blast cells or obvious dysplastic features was noted. These findings confirmed chronic PV with a normal karyotype, and bone marrow biopsy showed mild fibrosis.
He had been receiving continuous treatment with hydroxyurea. During follow-up every four months, leukocytosis, anemia, and thrombocytopenia developed in 2024 (Table 1). Peripheral blood smears showed 5.5% blasts, along with the pseudo-Pelger-Huët anomaly and degranulated neutrophils. Transformation to acute leukemia was suspected, and bone marrow examination was performed. The bone marrow showed hyperplasia, with cells at all stages of maturation in all three blood cell lines (Table 1).
In the granulocyte lineage, similar to the peripheral blood, the pseudo-Pelger-Huët anomaly and hypogranular neutrophils were observed, along with giant neutrophils with pink-colored cytoplasm and myeloperoxidase (MPO)-negative neutrophils. Eosinophils and basophils were also increased. MPO-positive blasts were identified in 18.8% of the cells. The blasts were medium to large in size with variable dimensions, with a nuclear-to-cytoplasmic ratio (N/C ratio) of 70-90%. The cytoplasm contained azurophilic granules and vacuoles. The nuclei exhibited partial clefts or indentations, with fine reticular chromatin and prominent nucleoli (Figure 1).
Bone marrow findings.(A) Blast with perinuclear hofs and cytoplasmic granules (closed arrow) and pseudo-Pelger-Huët anomaly and degranulated neutrophils (arrow head) (May-Giemsa staining, ×1,000). (B) Neutrophil with pink-colored cytoplasm (open arrow) (May-Giemsa staining, ×1,000). (C) Myeloperoxidase-positive blasts (closed arrow) and myeloperoxidase-deficient neutrophils (arrow head) (myeloperoxidase staining, ×1,000).
Flow cytometry (FCM) analysis showed the following phenotypic markers: CD4+, CD11c+, CD13+, CD19+, CD33+, CD34+, CD38+, CD117+, HLA-DR+, and cytoplasmic MPO+ (Figure 2).
Immunophenotypic findings of flowcytometry analysis.Blasts are positive for CD4, CD11c, CD13, CD19, CD33, CD34, CD38, CD117, HLA-DR, and cytoplasmic MPO.
The real-time quantitative polymerase chain reaction (RT-qPCR) detected RUNX1::RUNX1T1 at 1.1 × 10^4^ copies/μg RNA. No mutations in the FLT3 gene were detected. Chromosome analysis showed a karyotype of 46,XY,t(8;21)(q22;q22.1) in 18 of the 20 metaphases examined (Figure 3A). A computed tomography revealed mild splenomegaly. Based on the above findings, the patient was diagnosed with AML harboring the RUNX1::RUNX1T1 fusion gene, which had transformed from PV. JAK2V617F mutation analysis was performed in peripheral blood at the time of AML diagnosis using the i-densy IS-5320 (ARKRAY, Inc., Kyoto, Japan) [6] and showed JAK2V617F mutation positivity with a homozygous phenotype, and the allele burden was 85% or higher (Figure 3B).
Chromosomal analysis and JAK2V617F mutation analysis.(A) G-banding shows 46,XY,t(8;21)(q22;q22.1) (closed arrow). (B) JAK2V617F allele burden shows homozygous (wt, wild type; mt, mutant type). JAK2V617F mutation analysis was performed with peripheral blood obtained at the time of leukemia development as the material, using the i-densy IS-5320 (ARKRAY, Inc., Kyoto, Japan), and the melting curve analysis was performed using the genetic analysis support system, MEQNETiDia (ARKRAY, Inc.). The mutation rate of the JAK2 mutation was analyzed using the JAK2 mutation analysis software (ARKRAY, Inc.).
Induction therapy with the combination of venetoclax and azacitidine was administered. Venetoclax was shortened to a 21-day administration. After three courses of treatment, the patient demonstrated treatment resistance, leading to a decision to switch to best supportive care. The patient died approximately eight months after diagnosis of AML with RUNX1::RUNX1T1 fusion.
Discussion
While leukemic transformation and myelofibrosis are known endpoints of PV, other rare catastrophic complications can occur, such as spontaneous splenic rupture in CML. The present case exemplifies another rare event: transformation to core-binding factor AML. Both scenarios highlight the unpredictable nature of MPNs and the need for vigilance against diverse complications [7]. The overall survival of patients with PV has been reported to range from 13.5 to 24 years. PV is characterized by clinical features such as erythrocytosis, leukocytosis, thrombocytosis, and pruritus. Thrombotic events, MF following PV, and transformation to AML significantly impact the prognosis. The incidence of leukemia 20 years after a PV diagnosis ranges from 7.9% to 17%, with a particularly poor prognosis in cases that progress to leukemia. Risk factors for leukemia development after PV include older age, leukocytosis, venous thrombosis, and abnormal karyotype, among others [8,9]. Genomic profiles of PV and ET that transformed to leukemia indicate that mutations in genes such as TP53, TET2, RUNX1, ASXL1, and EZH2 are frequently associated with this outcome [10]. Regarding the evolution from PV and ET to AML, it is thought that the acquisition of genetic abnormalities leads to the development of leukemia, but the period from diagnosis to leukemic evolution is highly variable. The types of genetic mutations involved in short-term transformation and long-term transformation to AML appear to differ [10].
Interestingly, in de novo AML, balanced chromosomal translocations (PML::RARA, RUNX1::RUNX1T1, and CBFβ::MYH11) are frequently observed, whereas reports of such translocations in AML arising from MPN are scarce [4,11,12]. Furthermore, to the best of our knowledge, there is only one reported case of AML with the RUNX1::RUNX1T1 fusion gene that transformed from PV [13]. In the present case, cytogenetic analysis showed a t(8;21)(q22;q22.1) translocation, and RT-qPCR confirmed the presence of the RUNX1::RUNX1T1 fusion gene. A limitation of our report is the lack of next-generation sequencing data for mutations commonly associated with MPN transformation, such as TP53, ASXL1, and SRSF2. The presence of such additional mutations could provide further insight into the clonal evolution that led to the acquisition of the t(8;21) translocation. Therefore, it remains unclear whether the present case, like previously reported cases, represents a transformation to leukemia triggered by the acquisition of various genetic mutations as a long-term event of PV, or whether it represents an AML development mechanism similar to de novo AML. The patient had been taking hydroxyurea orally for over 20 years. Regarding the association between hydroxyurea and secondary AML in patients receiving long-term hydroxyurea therapy, the use of hydroxyurea alone has been reported to have a limited association with secondary AML [7,14,15]; however, the association between hydroxyurea and the transition of PV to AML with the RUNX1::RUNX1T1 fusion remains unknown.
Cases have been reported in which the JAK2V617F mutation disappears with the transformation to leukemia in patients with JAK2V617F-positive MPN [16,17]. In contrast, Asou et al. analyzed a case of transformation from ET to AML with RUNX1::RUNX1T1 fusion and found that the RUNX1::RUNX1T1 fusion gene was not detected in peripheral blood leukocytes after chemotherapy, but the JAK2V617F mutation was detected in both blasts before chemotherapy and peripheral blood leukocytes after chemotherapy; this suggested that the JAK2-mutated ET clones showed leukemic evolution [4]. In the present case, although analysis of JAK2V617F in AML cells was not performed, the high JAK2V617F allele burden (>85%) in peripheral blood at AML diagnosis strongly suggests that the leukemic blasts with the RUNX1::RUNX1T1 fusion evolved from the original JAK2-mutated PV clone, representing a subclone that acquired the t(8;21) translocation.
The morphological features of AML with the RUNX1::RUNX1T1 fusion gene are reported to be lacking in the erythroid and megakaryocytic lineages, but in the granulocytic lineage, characteristic findings include the pseudo-Pelger-Huët anomaly, degranulation, and pink-colored cytoplasm [18]. The blasts are large and vary in size, have a basophilic cytoplasm, and are characterized by numerous azurophilic granules, perinuclear hofs, and Auer rods [19]. In the present case, although Auer rods were not observed, the findings were consistent with those of AML harboring the RUNX1::RUNX1T1 fusion gene in many aspects. Flow cytometry analysis also showed the characteristic immune phenotype of expressing CD34 and CD19, in addition to myeloid markers [20], suggesting the presence of the RUNX1::RUNX1T1 fusion gene.
Hidalgo López et al. reported the bone marrow findings in 58 cases of PV with the blast phase, showing that 88% exhibited myeloid dysplasia, 72% had complex karyotypes, and 55% had TP53 mutation, with approximately 83% of the total cases diagnosed as AML myelodysplasia-related changes (MRCs) [21]. This finding suggests that, at the time of transformation to leukemia in MPN, including PV, the presence of genetic abnormalities or karyotype abnormalities, along with some form of morphological change, is a sign of PV and other MPNs transforming to leukemia. Therefore, follow-up observation of MPN should include not only blood counts and biochemical tests, but also morphological observations such as peripheral blood smears. Furthermore, given that follow-up in MPN may extend over a long period, as in the present case, capturing morphological changes in blood cells, such as the appearance of blasts or dysplasia, may serve as a helpful tool for understanding the disease process.
Conclusions
An extremely rare case of AML with the RUNX1::RUNX1T1 fusion transformed from JAK2-mutated PV was described in this report. PV progresses relatively slowly, but the prognosis is poor in cases that transform to MF or AML. In the transition phase from MPN to leukemia, detecting not only the appearance of leukemic blasts but also the emergence of morphological dysplasia can serve as a means to grasp the disease progression and provide opportunities for early intervention in treatment. Therefore, in the follow-up of patients with MPN, it is important to pay close attention not only to blood test data, but also to changes in blood cell morphology.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Polycythemia vera: 2024 update on diagnosis, risk-stratification, and management Am J Hematol Tefferi A Barbui T 146514879820233735795810.1002/ajh.27002 · doi ↗ · pubmed ↗
- 2The evolving genomic landscape of myeloproliferative neoplasms Hematology Am Soc Hematol Educ Program Nangalia J Green TR 287296201420142569686810.1182/asheducation-2014.1.287 · doi ↗ · pubmed ↗
- 3Genetic characterization and risk stratification of acute myeloid leukemia Cancer Manag Res Pourrajab F Zare-Khormizi MR Hashemi AS Hekmatimoghaddam S 223122531220203227376210.2147/CMAR.S 242479 PMC 7104087 · doi ↗ · pubmed ↗
- 4Transformation into acute myeloid leukemia with t(8;21)(q 22;q 22.1); RUNX 1::RUNX 1T 1 from JAK 2-mutated essential thrombocythemia: a case report J Med Case Rep Asou C Sakamoto T Suzuki K 3721820243915417010.1186/s 13256-024-04691-0PMC 11330597 · doi ↗ · pubmed ↗
- 5Evaluation of diagnostic criteria in polycythemia vera Semin Hematol Pearson TC 212438200110.1016/s 0037-1963(01)90136-211242598 · doi ↗ · pubmed ↗
- 6Simultaneous detection of JAK 2, CALR, and MPL mutations and quantitation of JAK 2 V 617F allele burden in myeloproliferative neoplasms using the quenching probe-Tm method in i-densy IS-5320 Int J Lab Hematol Arai K Sakaguchi M Yui S 110211104420223603979510.1111/ijlh.13938 · doi ↗ · pubmed ↗
- 7An autopsy presentation of spontaneous splenic rupture in chronic myeloid leukemia: a rare case report J Med Surg Public Health Kanani J Sheikh MI 10011832024
- 8Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors Blood Cancer J Cerquozzi S Tefferi A 05201510.1038/bcj.2015.95PMC 467094826565403 · doi ↗ · pubmed ↗