Force-Triggered Thermodynamically Uphill Disulfide Reduction through Sulfur Oxidation State Control
Marc Mora, Georgia Cohen, William Cranton, Olaia Anton, Amy E. M. Beedle, Guillaume Stirnemann, Sergi Garcia-Manyes

TL;DR
This study shows that mechanical force can enable a disulfide bond in a protein to be reduced by inorganic oxyanions, which is not possible under normal thermodynamic conditions.
Contribution
The paper demonstrates that mechanical forces can activate disulfide reduction by inorganic oxyanions, which is thermodynamically uphill.
Findings
Mechanical force activates disulfide reduction by inorganic oxyanions in proteins.
This force-induced reaction affects protein elasticity and function.
DFT calculations and single-molecule experiments support the force-triggered mechanism.
Abstract
In addition to thermal energy, current, and light, mechanical forces activate chemical reactions, often steering reaction pathways that result in products different from those obtained under thermodynamic control. Single-molecule mechanochemistry experiments have probed how the forced activation of a single covalent bond results in accelerated scission of both homolytic and heterolytic bonds, and the ring-opening of strained mechanophores in long polymers. Due to its mechanistic simplicity, the concerted SN2 thiol–disulfide nucleophilic substitution has been successfully used as a model system to interrogate how the nucleophilicity of an attacking organic, low-oxidation state thiol determines the force dependency of the thiol/disulfide exchange rate. Inorganic sulfur-oxyanions are comparatively much less reactive. Whether mechanical forces can activate the rupture of a protein disulfide…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1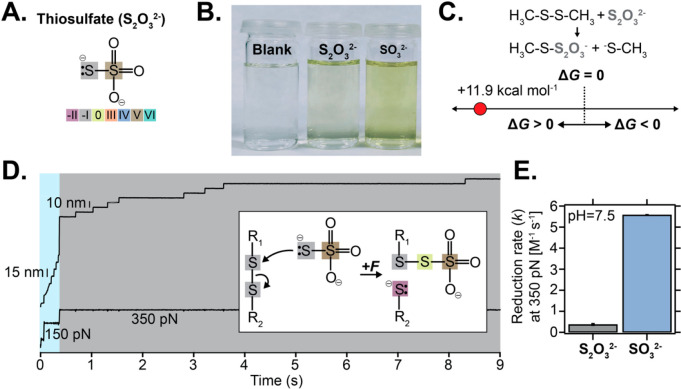 2
2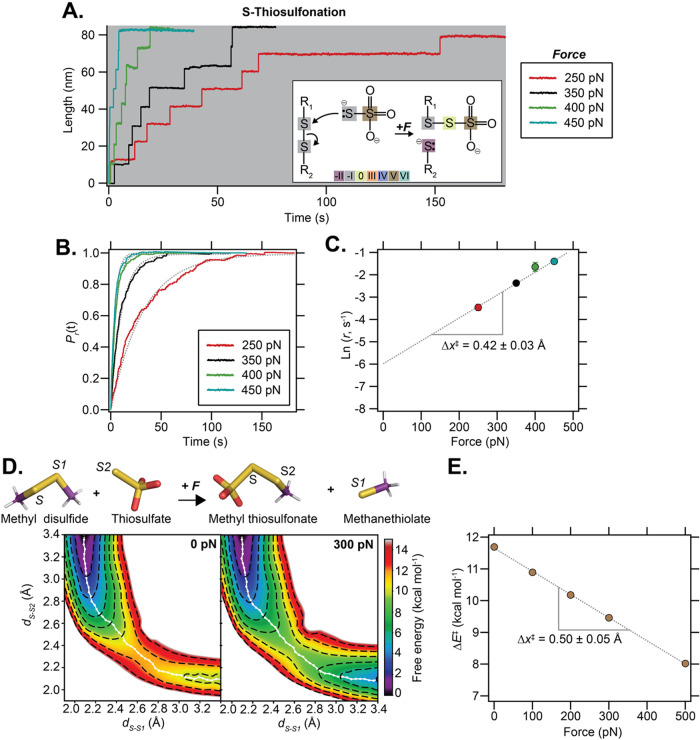 3
3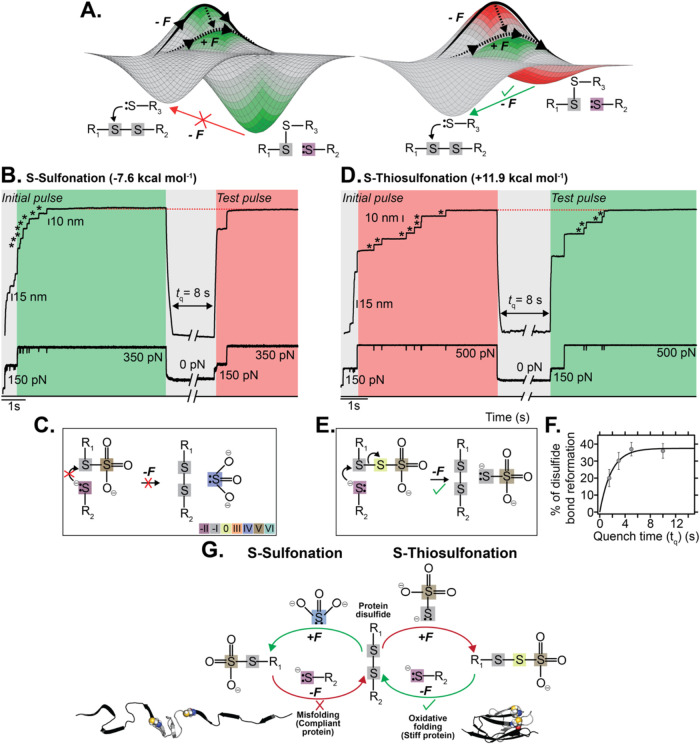 4
4- —Wellcome Trust10.13039/100010269
- —Wellcome Trust10.13039/100010269
- —Medical Research Council10.13039/501100000265
- —Biotechnology and Biological Sciences Research Council10.13039/501100000268
- —Leverhulme Trust10.13039/501100000275
- —Leverhulme Trust10.13039/501100000275
- —Royal Society10.13039/501100000288
- —Cancer Research UK10.13039/501100000289
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsThermal and Kinetic Analysis · Electrochemical Analysis and Applications · Metal Extraction and Bioleaching
Introduction
Mechanical force provides an energy source, alternative to heat, electric current and light, to initiate a chemical reaction.? Specifically, externally applied force can bias the potential energy surface of chemical reactions, favoring routes to products that are not necessarily populated according to a Boltzmann distribution of energy barriers.? For example, the application of forces through ultrasounds catalyzed covalent ring-opening reactions in mechanophores contained in long polymer chains? and biased reaction pathways toward the formation of thermodynamically disfavored isomers.? In some instances, reaching low-probability products resulted from visiting transition state structures the lifetime of which was regulated by force.?
Single-molecule experiments have provided detailed mechanistic understanding of the localized effect of force on the activation of a specific covalent bond? for a variety of homolytic and heterolytic reactions.? In particular, force-clamp experiments demonstrated how the bimolecular nucleophilic substitution leading to disulfide rupture? is accelerated by force, ?,? and showed how the chemical nature of the attacking nucleophile regulates the rate of disulfide reduction. ?−? ? For the specific case of organic thiols (−II sulfur oxidation state), a direct correlation between the electrostatic partial charge localized on the nucleophilic sulfur and the measured disulfide reduction rate was obtained. ?,? However, a systematic understanding of how force modulates the rate and outcome of bimolecular chemical reactions that encompass a broad range of oxidation states is missing.
Due to its unique electronic configuration, displaying an empty external d-orbital available for bonding ([Ne]3s^2^3p^4^3d^0^), sulfur exhibits versatile reactivity with oxidation states ranging from −II to +VI. Hence, inorganic sulfur nucleophiles, exhibiting a broad range of oxidation states, provide an excellent platform to interrogate how force modulates the underlying one-dimensional (1D) energy landscape of S_N_2 reactions, potentially affecting both their kinetics and thermodynamics.
Results
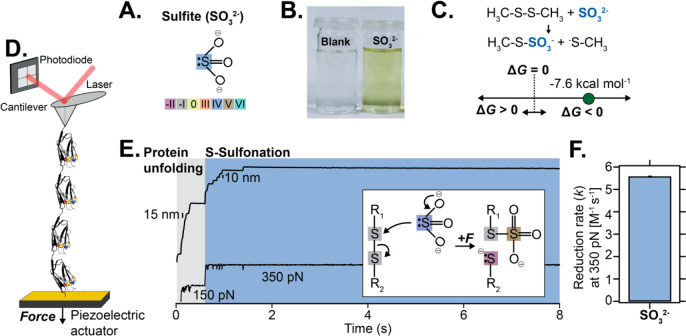
We started by testing the reactivity of sulfite (SO_3_ ^2–^), which exhibits a +IV sulfur oxidation state (FigureA). In the absence of force, sulfite reduced the symmetric disulfide bridge of the Ellman’s reagent? (FigureB). These findings were corroborated with density functional theory (DFT) calculations (using the M06–2X? functional with the ma-def2-TZVP basis set? in CPCM implicit solvent) to predict the standard Gibbs free energy (ΔG ^0^) associated with the cleavage of the disulfide bond between two model thiols (here, methyl thiols) by sulfite, displaying a favorable tendency toward disulfide bridge reduction (ΔG ^0^ = −7.6 kcal mol^–1^), FigureC. From the kinetic perspective, however, the localized charge on the sulfur estimated based on the DFT calculations is only slightly negative (−0.055 e, Figure S1)suggesting a much slower reactivity than that measured for organic (−II) thiols, for which we could previously establish a correlation between the rate constant and the sulfur charge.? A different combination of DFT functional and basis set gave the same qualitative results (Figure S1). While recent machine-learned reactive potentials? applied to chemical reactivity? would provide a more quantitative assessment of disulfide bond reactivity toward these nucleophiles (for example by taking into account full explicit solvation and the presence of ions), our admittedly simpler calculations already capture our experimental observations.
Force accelerates the thermodynamically spontaneous reduction of a protein disulfide bond by sulfite anion. (A) Sulfite anion and the sulfur oxidation state. (B) Sulfite disrupts the symmetric disulfide bridge of the Ellman’s reagent (A λ=412 nm = 1.058 au). (C) DFT calculations of the standard free energy (M06–2X functional with the ma-def2-TZVP basis set in CPCM implicit solvent) associated with disulfide rupture of two methyl thiols by sulfite. (D) Schematic representation of the single-molecule force-clamp experiment, where an individual (Ig27E24C–K55C)8 polyprotein is stretched in an AFM. (E) Force application first triggers the unfolding of the protein up to the disulfide bond (gray region). Sulfite (250 mM, pH 7.5) induces the rupture of the stretched disulfide bond, marked by a ∼10 nm stepwise increase in the protein length (blue region). Inset: schematics of the sulfite-mediated disulfide rupture. (F) The concentration-normalized rate of protein disulfide rupture by sulfite at a constant force (F = 350 pN), pH 7.5; N = 15 reduction trajectories.
We then questioned whether mechanical force might accelerate the sulfite-mediated rupture of an individual disulfide bond. We employed a single-molecule force-clamp AFM assay designed earlier ?,?,? using a polyprotein containing eight repeats of a titin Ig27 mutant with a buried engineered disulfide bridge between positions 24 and 55, E24C–K55C (FigureD). Briefly, the application of a constant force (150 pN for 0.5 s) promoted the unfolding of the protein domain(s) up to the rigid disulfide bond, hallmarked by a ∼15 nm stepwise increase of the protein (FigureE). Each unfolding event readily exposes the previously cryptic disulfide bridge to the solution. Subsequently, the application of a higher force (F = 350 pN) in the presence of 250 mM sulfite at pH 7.5 triggered the cleavage of the disulfide bridge, fingerprinted by individual ∼10 nm steps that correspond to the length release of the amino acids previously trapped by the covalent disulfide bond. Averaging a number of independent reduction trajectories normalized by the sulfite concentration yielded a reduction rate of k = 5.58 M^–1^ s^–1^ (FigureF), which is certainly much slower than that measured for organic thiols with lower (−II) oxidation state,? even though sulfite nucleophilicity (and the overall reaction thermodynamics) is modulated by its protonation state (pk a = 6.97) (Figures S2–S4). Our results highlight that force can significantly accelerate the otherwise kinetically slow (yet thermodynamically favored) S_N_2 cleavage of a protein disulfide bond by sulfite.
We then queried whether a structurally similar compound such as thiosulfate (S_2_O_3_ ^2–^) (FigureA), with a nucleophilic sulfur atom with a lower oxidation state (−I)? exhibits different reactivity toward disulfide reduction under force. Critically, both the Ellman’s assay (FigureB) and DFT calculations (ΔG ^0^ = +11.9 kcal mol^–1^) (FigureC) concluded that disulfide bond reduction by thiosulfate is thermodynamically nonfavored (which is qualitatively confirmed using another combination of DFT functional/basis set, Figure S5). We therefore questioned if the application of mechanical force would enable disulfide bond cleavage, even in these nonfavorable thermodynamic conditions. Our force-clamp experiments (FigureD) revealed that, albeit exhibiting a slower normalized rate at 350 pN of disulfide reduction (k = 0.38 M^–1^ s^–1^) than sulfite, thiosulfate (250 mM, pH 7.5) can indeed induce the nonspontaneous rupture of a disulfide bond that is exposed to a persistent and calibrated stretching force (FigureE). To elucidate how the kinetics of disulfide bond scission depends on the pulling force, we measured the time-course of disulfide bond rupture at several forces spanning 250 pN–450 pN (FigureA). To obtain the rate of reduction at any particular force, we averaged and normalized 16–40 individual traces, such as those shown in FigureA, and fitted the average trajectory to a single exponential (dotted lines in FigureB). From the exponential time constant of the fits (τ_R_) we calculated the rate of reduction as r = 1/τ_R_. The standard error of the mean (s.e.m.) of these data was estimated by bootstrapping. We then fitted the force dependency of the rate of reduction using the simple Arrhenius term r(F) = r _ o _exp(FΔx ^‡^/kT), where r 0 is the rate constant in the absence of force,? yielding r 0 = 0.0025 ± 0.0007 s^–1^ and Δx ^‡^ = 0.42 ± 0.03 Å (FigureC). These results conclude that force accelerates the rate of disulfide bond reduction in the presence of a thermodynamically nonfavored nucleophile such as thiosulfate.
Mechanical force induces the thermodynamically impaired reduction of a protein disulfide bond by thiosulfate anion. (A) Chemical representation of thiosulfate anion. (B) Thiosulfate can barely react with the Ellman’s reagent, as opposed to sulfite (A λ=412 nm = 0.270 au and 1.058 au, respectively). (C) DFT calculations of the standard free energy (M06–2X functional with the ma-def2-TZVP basis set in CPCM implicit solvent) associated with disulfide rupture of two methyl thiols by thiosulfate. (D) Kinetics of disulfide bond reduction at 350 pN in the presence of 250 mM of thiosulfate (pH 7.5). Inset: schematics of the underpinning chemical reactivity. (E) The concentration-normalized rate of protein disulfide rupture by thiosulfate (gray) and sulfite (blue, for comparison) at a constant force (F = 350 pN), pH 7.5; N = 34 trajectories.
*The kinetics of S-thiosulfonation under force. (A) The rate of disulfide bond reduction by thiosulfate (250 nM at pH 7.5) is accelerated by the stretching force, as observed during the second force pulse (Figure D) at pulling forces ranging from 250 pN to 450 pN. (B) Measurement of the rate of disulfide bond reduction by thiosulfate at four different pulling forces. At each force, we averaged and normalized 15–40 traces to obtain the probability of reduction (P
r ). The reduction rate at each force is obtained by fitting a single exponential to the average trace (dotted gray line). 250 pN N = 25 trajectories; 350 pN N = 34 trajectories; 400 pN N = 20 trajectories; 450 pN N = 16 trajectories. (C) Semilogarithmic plot of the disulfide bond reduction rate by thiosulfate as a function of the pulling force. Fits of the Arrhenius term r(F) = r 0 exp(FΔx ‡/kT) to the experimental data give Δx ‡ = 0.42 ± 0.03 Å (dotted gray line). (D) Energy surface for the reaction between methyldisulfide and thiosulfate obtained from quantum calculations at 0 pN (left), to which the work of force on d S–S1 was added to obtain the same surface at increasing forces, for example 300 pN (right). The white string corresponds to the average minimum energy path at 300 K. Atom labels are shown on the molecular structures shown above, which correspond to the minimized conformations in the reactant and product states, respectively. (E) The reaction energy barrier is determined at each force as the difference between the maximum value of the energy along the path and that of the first point of the path (reactant state, upper left). The force dependency allows determination of the distance to the transition state in the Bell model formalism.*
To obtain an atomistic, detailed picture of how force modulates the free-energy surface of disulfide bond scission by thiosulfate, we complemented our single bond experiments with DFT calculations. These calculations enabled us to identify the height of the main energy barrier determining the reaction (ΔE 0 ^‡^) and measure how its height is modulated by the applied force (FigureD). ΔE 0 ^‡^ was determined by a systematic scan along the initial disulfide bond distance (d SS) and sulfur-nucleophile distance, which allowed us to identify the lowest energy pathway between reactants and products, and consequently the barrier (Figure S6). The energy landscape under force was then obtained by adding a work –Fd SS contribution and then using the protocol described above to determine the force-dependent barrier ΔE ^‡^(F). Assuming a simple Bell/Arrhenius behavior, ΔE ^‡^(F) = ΔE 0 ^‡^ – FΔx ^‡^, these calculations resulted in a Δx ^‡^ = 0.50 ± 0.05 Å (FigureE).
Alternatively, Δx ^‡^ can be independently calculated by projecting the energy path along d SS_1 _ and measuring the difference in d SS_1 _ between reactant and product. As the top of the barrier is relatively flat, the measured Δx ^‡^ slightly depends in this case on the pulling force, rapidly plateauing toward 0.51 Å (Figure S7). In both cases, the excellent agreement between the calculated and the experimental Δx ^‡^ values suggest that, similar to organic thiols, the distance to the transition state of the reaction can be related to the elongation of the S–S bond at the transition state.?
We next focused on the reversibility of the force-induced reaction. We conjectured, using a similar interpretation to that employed to rationalize covalent S_N_2 disulfide bond rupture with organic nucleophiles,? that reaction activation occurs once force lowers the energy barrier that separates products from reactants in the 1D reaction coordinate probed in the single-molecule experiments (FigureA). For a thermodynamically favored reaction, products have a lower free energy than reactants (ΔG ^0^ < 0, green region) (FigureA, left), and the application of force speeds up barrier crossing. However, returning products back to reactants in the absence of force is uphill, implying that the reverse reaction does not occur (red arrow). On the contrary, for a thermodynamically nonfavored reaction, the free energy of the products is higher than that of the reactants (ΔG ^0^ > 0) and the barrier separating both states is large (red region). In this limit case, force can lower the energy barrier by an amount such that the barrier cusp aligns with the free energy of the products (green region), in which case the reaction becomes suddenly possible (FigureA, right). The corollary is that, upon force withdrawal, the initially nonfavored products can suddenly transform back into reactants spontaneously (green arrow).
The reversibility of force-induced S-thiosulfonation. (A) Schematics of a thermodynamically favored (left) and disfavored (right) disulfide SN2 exchange energy surface under force. (B) Protein disulfide bond reduction by sulfite is thermodynamically favored (green region), resulting in a low-energy mixed-disulfide product that cannot reversibly reform the initial disulfide reactant in the absence of force. This is marked by the absence of ∼10 nm steps in the test pulse (red region, N = 18 trajectories) after a force quenching time t q = 8 s. Dotted red line marks the full extension of the protein after the Initial and Test pulses. (C) Schematics of the reactivity defining the irreversible S-sulfonation. (D) Protein disulfide bond reduction by thiosulfate, while thermodynamically not favored (red region), is possible under force. Such a force-catalyzed reactivity renders the reaction suddenly reversible, hallmarked by the presence of ∼10 nm steps in the refolding pulse (green region) after a t q = 8 s. (E) Schematics of the reversible S-thiosulfonation. (F) The percentage of protein disulfide bond reformation increases exponentially with t q, yielding a bond reformation rate k = 0.54 s–1 (N 1.5s = 10, N 3s = 23, N 5s = 28 and N 10s = 24 trajectories). (G) Summary of the implications of chemical reactivity on protein nanomechanics.
We employed our single-molecule force-quench experiments to test both potentially distinct reversibility scenarios when an individual protein disulfide bond is reduced by either the thermodynamically favored sulfite (SO_3_ ^2–^) or the thermodynamically impaired thiosulfate (S_2_O_3_ ^2–^). Using a five-force-pulse protocol (FigureB), we first applied an initial short pulse of 150 pN to the (Ig27_E24C–K55C_)8 polyprotein, resulting in the unfolding of the protein up to the disulfide bridge, which becomes solvent-exposed. A second pulse at a higher force (350 pN) triggered a ∼10 nm stepwise increase in protein length (asterisks), corresponding to the force-mediated rupture of each individual disulfide bond by the attacking sulfite. The initial two-force pulse results in a fully stretched and reduced protein containing a free cysteine thiolate and a mixed disulfide between the protein cysteine and the attacking sulfite, thus creating an S-sulfonated protein. Removal of the stretching force for t q = 8 s triggered the collapse of the protein, potentially enabling its oxidative folding. A second test pulse, mirroring the initial pulse, probed the chemical and folding reversibility success for a given t q. The subsequent reapplication of force resulted in a protein extension devoid of any steps for any t q (FigureB). The lack of ∼10 nm steps in the test pulse unambiguously demonstrated that the mixed-disulfide between sulfite and the protein cysteine is energetically very stable, thus preventing the reattack by the free protein thiols (FigureC), consequently inhibiting refolding. These results are in line with DFT calculations and the colorimetric Ellman’s assay; since disulfide reduction by sulfite is thermodynamically favored, the reverse reaction is necessarily impaired.
We then repeated the same refolding experiments in the presence of thiosulfate. In this case, analogous to the initial force-pulse exhibiting the unfolding and the (thermodynamically impaired) reduction of the disulfide bond under force (FigureD), the elongation of the protein in the test pulse followed a sequential reduction of individual disulfide bonds (∼10 nm steps) (FigureD). Of note, the efficiency of disulfide bond reformation depends on the time the force is quenched, t q. In this particular trajectory, the Initial pulse (red) exhibits 7 individual disulfide bond rupture events (marked with an asterisk), whereas the Test pulse (green) contains only 4 events. This implies that, for t q = 8 s, not all disulfide bonds completely reformed, but rather 4/7 = 57%. Crucially, the presence of disulfide rupture events in the Test pulse fingerprints the reversibility of the S-thiosulfonation (FigureE). The probability of reforming the protein disulfide depends on the quenching time (t q) (FigureF), ultimately resulting in a successfully refolded (and reoxidized) protein. Also, in this case, the reaction reversibility can be predicted by DFT calculations. Given that disulfide reduction by thiosulfate is not thermodynamically favored (and nevertheless activated by force), the reverse reaction (i.e., the reformation of the disulfide bond from the S-thiosulfonated moiety and the neighboring thiolate) will be necessarily spontaneous, in line with our single-molecule observations.
Discussion
We primarily used the Ig27 protein as a well-defined substrate to interrogate chemical reactivity under force. However, given the protein context where chemical reactivity takes place, our conclusions have direct implications for the relationship between (nonenzymatic) oxidative folding and protein elasticity. When post-translationally sulfonated after sulfitolysis,? the mechanically stiff Ig27 titin domain turns into a compliant polypeptide devoid of mechanical stability. By contrast, upon S-thiosulfonation under force, and in the presence of a neighboring thiol in the protein structure, the mechanically stiff disulfide bonds are readily reformed, altogether rendering the Ig27 protein properly refolded and mechanically stable (FigureG). Consequently, disulfide cleavage with two structurally and chemically similar oxyanions gives rise to two protein forms of markedly different mechanical functions. Given the emerging role of sulfite and thiosulfate in cardiac and mitochondrial dysfunction and adverse reactions,? and in aortic smooth muscle cells differentiation and stimulation of angiogenesis and vascular repair,? respectively, it is tempting to hypothesize that their related post-translational modifications might be related to the gain and loss of mechanical function of the proteins at play.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Garcia-Manyes S.Beedle A. E. M.Steering chemical reactions with force Nat. Rev. Chem.2017111008310.1038/s 41570-017-0083 · doi ↗
- 2Liu Y.Holm S.Meisner J.Jia Y.Wu Q.Woods T. J.Martinez T. J.Moore J. S.Flyby reaction trajectories: Chemical dynamics under extrinsic force Science 2021373655120821210.1126/science.abi 760934244412 · doi ↗ · pubmed ↗
- 3Davis D. A.Hamilton A.Yang J.Cremar L. D.Van Gough D.Potisek S. L.Ong M. T.Braun P. V.Martinez T. J.White S. R.Force-induced activation of covalent bonds in mechanoresponsive polymeric materials Nature 20094597243687210.1038/nature 0797019424152 · doi ↗ · pubmed ↗
- 4a Hickenboth C. R.Moore J. S.White S. R.Sottos N. R.Baudry J.Wilson S. R.Biasing reaction pathways with mechanical force Nature 2007446713442342710.1038/nature 0568117377579 · doi ↗ · pubmed ↗
- 5Lenhardt J. M.Ong M. T.Choe R.Evenhuis C. R.Martinez T. J.Craig S. L.Trapping a diradical transition state by mechanochemical polymer extension Science 201032959951057106010.1126/science.119341220798315 · doi ↗ · pubmed ↗
- 6Beyer M. K.Clausen-Schaumann H.Mechanochemistry: the mechanical activation of covalent bonds Chem. Rev.200510582921294810.1021/cr 030697 h 16092823 · doi ↗ · pubmed ↗
- 7a Zheng P.Li H.Highly covalent ferric-thiolate bonds exhibit surprisingly low mechanical stability J. Am. Chem. Soc.2011133176791679810.1021/ja 200715 h 21476573 · doi ↗ · pubmed ↗
- 8a Nagy P.Kinetics and Mechanisms of Thiol-Disulfide Exchange Covering Direct Substitution and Thiol Oxidation-Mediated Pathways Antioxid. Redox Signaling 201318131623164110.1089/ars.2012.4973 PMC 361317323075118 · doi ↗ · pubmed ↗