Potential immunogenic modulation of hypo-fractionated radiotherapy at optimal schedules and the subsequent vaccine-like effect of local irradiation - a systematic review
Jagtar Singh, Martin Ashdown, Siddhartha Baxi

TL;DR
This systematic review explores how hypo-fractionated radiotherapy can activate the immune system and potentially act like a vaccine to fight cancer.
Contribution
The paper systematically reviews the immunogenic effects of optimal hypo-fractionated radiotherapy schedules and their vaccine-like potential.
Findings
HFRT with optimal regimens activates T lymphocytes and reduces Tregs.
T-cell infiltration into tumors was observed in most studies after HFRT.
Optimal HFRT schedules may induce immune responses and abscopal effects.
Abstract
Hypo-fractionated radiotherapy (HFRT) regimens can induce immune system activation and help to identify a therapeutic window after RT by measuring cytotoxic T-cell concentration. Here, we summarise previous preclinical and clinical studies on the effects of HFRT on the immune system, both locally and systemically. We also investigate the existing data on the optimal dose and fractionation scheme of HFRT to enhance local and distant anti-tumour immunity. A search was conducted using the PubMed, ScienceDirect, and Google Scholar databases. The systematic review was conducted in accordance with the PRISMA-DTA guidelines. Quality was assessed utilising the Prediction model Risk Of Bias ASsessment Tool (PROBAST). Data from publications that met quality criteria were grouped via (1) hypo-fractionated radiotherapy, (2) CD8+ T-cells infiltration, (3), immune stimulation, and (4) abscopal…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
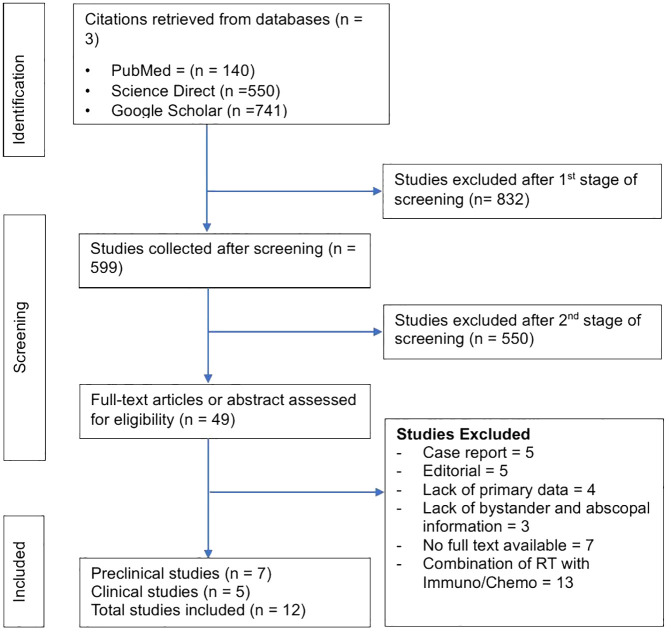 Figure 1
Figure 1| Search strategy | |
|---|---|
| • PubMed | |
| Academic databases searched | • Science Direct |
| • Google Scholar | |
| • Journals papers | |
| Targeted items | • Workshop papers |
| • Conference papers | |
| • Non-academic papers | |
| • Titles | |
| Searched applied to | • Abstracts |
| • Keywords (RT, hypo-fractionation RT, immune system, anti- | |
| Language | • English |
| Publication periods | • Published between 2010 and 2024 |
| • RT-induced immune stimulation | |
| Outcomes | • Improves bystander and Abscopal effects |
| • Clinical outcomes, such as treatments and immune-related | |
| Authors, year | Pat. no. | Tumour type | Biopsy sample | HFRT schedule/no. fractions | Total dose (Gy) |
|---|---|---|---|---|---|
| Preclinical studies | |||||
| Filatenkov et al. (2015) ( | 14 mice | Colon tumours | CT26 and MC38 Cell lines | 30 Gy × 1 fr | 30 Gy |
| Markovsky et al. (2019) ( | NA | Breast cancer and | 67NR murine and | 3.5 Gy/minute | 10 Gy |
| Kim et al. (2023) ( | NA | Lung metastasis | FSaII, CT-26, and 4T1 cells | 20 Gy × 1 fr and | 20 Gy |
| Frey et al. (2017) ( | NA | Colon Cancer | CT26 cells | 5 Gy × 2 frs | 10 Gy |
| Zhao et al. (2022) ( | NA | Lung Cancer | Lewis lung carcinoma (LLC) cells | 3.7 Gy × 4 frs, | 20Gy |
| Schaue et al. (2012) ( | NA | Melanoma | B16-OVA murine | 15 Gy × 1 fr and | 15 Gy |
| Grapin et al. (2019) ( | NA | Colon Cancer | CT26 cells | 2 Gy × 18 frs | 36 Gy |
| Clinical studies | |||||
| Zhang et al. (2017) ( | 6 | Non-small-cell lung cancer | Blood | 48 Gy × 8 frs or | 48 Gy |
| Muraro et al. (2017) ( | 21 | Breast Cancer | Blood | 10 Gy × 3 frs | 30 Gy |
| McGee et al. (2018) ( | 31 | Lung, | PBMC and serum (pre- and 1–2 weeks post-SAR) | 1–5 frs SBRT or | NA |
| Tubin et al. (2019) ( | 23 | Lung = 16 | NA | 10–12 Gy × 1–3 frs | 10–12 Gy |
| Tubin et al. (2019) ( | 60 | Non-small cell lung cancer | NA | 10 Gy × 3 frs | 30 Gy |
| Authors, year | Country | Sample | Treatment option | RT dose/no. fractions |
| T-cell response observed |
|---|---|---|---|---|---|---|
| Preclinical studies | ||||||
| Filatenkov et al. (2015) ( | USA | CT26 and MC38 Cell lines | SABR | 30 Gy × 1 fr | NA | Yes |
| Markovsky et al. (2019) ( | USA | 67NR murine, | SFR | 10Gy × 3.5 Gy/minute | NA | Yes |
| Kim et al. (2023) ( | Korea | FSaII, CT-26, and 4T1 cells | HFRT | 20 Gy × 1 fr and | NA | Yes |
| Frey et al. (2017) ( | Germany | CT26 cells | HFRT | 5 Gy × 2 frs | NA | Yes |
| Zhao et al. (2022) ( | China | Lewis lung carcinoma (LLC) cells | HFRT | 3.7 Gy × 4 frs, |
| Yes |
| Schaue et al. (2012) ( | USA | B16-OVA murine | SFR | 15 Gy × 1 fr and | NA | Yes |
| Grapin et al. (2019) ( | France | CT26 cells | HFRT | 2 Gy × 18 frs |
| Yes |
| Clinical studies | ||||||
| Zhang et al. (2017) ( | China | Blood | SBRT | 48 Gy × 8 frs or |
| Yes |
| Muraro et al. (2017) ( | Italy | Blood | SBRT | 10 Gy × 3 frs | NA | Yes |
| McGee et al. (2018) ( | USA | PBMC and serum (pre- and 1–2 weeks post-SAR) | SAR | 1–5 frs SBRT or | NA | Yes |
| Authors and Year | Country | Sample | Study endpoint |
| Tumour control observed |
|---|---|---|---|---|---|
| Preclinical studies | |||||
| Kim et al. (2023) ( | Korea | FSaII, CT-26, and 4T1 cells | Tumour control | One-day interval RT (P = 0.641), | Yes |
| Frey et al. (2017) ( | Germany | CT26 cells | Tumour control | NA | Yes |
| Zhao et al. (2022) ( | China | Lewis lung carcinoma (LLC) cells | Tumour control |
| Yes |
| Schaue et al. (2012) ( | USA | B16-OVA murine | Tumour control | NA | Yes |
| Grapin et al. (2019) ( | France | CT26 cells | Tumour control | NA | Yes, |
| Clinical studies | |||||
| Tubin et al. (2019) ( | Austria | NA | Tumour control | NA | Yes |
| Authors and Year | Country | Sample | Treatment option | Study endpoints | P - values | Response rate observed |
|---|---|---|---|---|---|---|
| Filatenkov et al. (2015) ( | USA | CT26 and MC38 Cell lines | SABR | CR | NA | Yes |
| Markovsky et al. (2019) ( | USA | 67NR murine, Lung Carcinoma (LLC) mouse model | SFR | AE | NA | Yes |
| Zhao et al. (2022) ( | China | Lewis lung carcinoma (LLC) cells | HFRT | OS | NA | Yes |
| Clinical studies | ||||||
| Zhang et al. (2017) ( | China | Blood | SBRT | OS | NA | Yes |
| Muraro et al. (2017) ( | Italy | Blood | SBRT | PFS | NA | Yes |
| McGee et al. (2018) ( | USA | PBMC and serum (pre- and 1–2 weeks post-SAR) | SAR | AE | NA | Yes |
| Tubin et al. (2019) ( | Austria | NA | SBRT | OS |
| Yes |
| Tubin et al. (2019) ( | Austria | NA | SBRT | BE | NA | Yes |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRadiation Therapy and Dosimetry · Advanced Radiotherapy Techniques · Effects of Radiation Exposure
Introduction
1
Radiation therapy (RT) is usually considered a “local” treatment modality in cancer therapy because radiation can only directly eradicate cancer cells within the radiation field. Because of recent developments in image guidance and RT delivery methods, single ablative high doses can be safely delivered to many tumour sites by using stereotactic radiosurgery (SRS), stereotactic body RT (SBRT), or stereotactic ablative body irradiation (SABR) (1–4). High doses of radiation can be achieved by a single treatment (extreme oligo-fractionation) or by 2 to 5 high-dose treatments (oligo-fractionation or hypo-fractionation), serving as an alternative to conventional daily low-dose fractionated treatments (<3 Gy) over several weeks (5). Limited results showed improved efficacy compared with traditional fractionated RT in managing advanced or metastatic colorectal, liver, and non-small cell lung tumours (2). The outcome can be comparable to surgery for resectable tumours, and SRS can be applied to unresectable tumours (2, 6).
Hypo-fractionated RT (HFRT) is a modern radiation technique that provides targeted high-dose irradiation to a tumour while limiting damage to surrounding normal tissues (7–9). HFRT directly kills tumour cells via DNA double-strand breaks and propagates dose-dependent vascular damage and destruction of the tumour microenvironment, causing secondary tumour cell death (10–12). Massive tumour cell death because of DNA damage and vascular injury functions can produce strong anti-tumour immunity. Therefore, it has been reported that the anti-tumour immune response plays a significant role in the outcome of SABR (10–12). However, RT may result in poor outcomes in patients with a weakened immune system, whereas it may effectively eradicate tumours in patients with a more robust immune system (13, 14).
It has been shown that RT may contribute to making tumours visible to the immune system (15–19). After RT treatment, MHC-I molecules display an increased pool of peptides for antigen presentation (20). Dendritic cells (DCs) can capture tumour-associated antigens (TAAs) released to the tumour periphery (21). These DCs become active via toll-like receptor (TLRs) recognition, in which endogenous danger signals emitted by dying tumour cells are identified (21). The activation of DCs is characterised by the upregulation of cell surface molecules involved in antigen presentation and co-stimulation (e.g., CD80 and CD86) and the release of pro-inflammatory cytokines (21). Thus, activated DCs migrate to secondary lymphoid organs, where TAAs are presented to CD4+ Th cells in the MHC-II context (21). Active, effector T-cells may recirculate through the body and generate a tumour-specific immune response in distant areas (21). Using this mechanism, adaptive immune responses may help to eradicate metastasis of tumours that do not express MHC-II. CD4+ T-cells may help kill tumour cells through several mechanisms (21). One such mechanism enables the development of tumour-specific CD8+ T-cells that recognise tumour peptides by MHC-I (21).
A growing body of evidence suggests that the systemic anti-tumour effect in metastatic disease in response to high-dose local radiation results in the regression of non-irradiated distant tumour sites (22). This phenomenon, known as the abscopal effect (AE) of radiation, was first described by RH Mole in 1953 (23). Multiple mechanisms have been proposed to cause the AE (16, 24), such as the systemic secretion of specific cytokines and chemokines, a systemic immune response against local tumour antigens released, or local inflammation that can lead to a distant effect (25). In any case, the hypothesis that the AE is immune-mediated is becoming stronger. If the radiation dose is sufficient to generate cell death, this can lead to the induction of the adaptive immune response. RT directly elicits an innate immune recognition of tumour by releasing danger signals”. Thus, these signals can increase immune-mediated cell death, which promotes the uptake of circulating tumour antigens by DCs via cross-priming and ultimately leads to the activation of tumour-specific T-cell response (26). The tumour-specific T-cells are capable of recirculating throughout the body, detecting any tumour cells (across multiple antigens) and eradicating them (24, 27). Therefore, tumours that are even at a distance from the irradiated field can be immunologically killed (24, 27). This is described as an AE (24, 27, 28).
Here, we summarise previous preclinical and clinical studies on the effects of hypo-fractionated RT (HFRT) on the immune system locally and systemically. We also investigate the existing data on the optimal dose and fractionation scheme of HFRT to enhance local and distant anti-tumour immunity.
Materials and methods
2
Search strategy and study selection
2.1
This systematic review followed the PRISMA statement for reporting systematic reviews and meta-analyses (29). A comprehensive electronic search was conducted between March and October 2024 using PubMed (https://pubmed.ncbi.nlm.nih.gov/), ScienceDirect (https://www.sciencedirect.com/), and Google Scholar (https://scholar.google.com.au/) databases for articles published between 2010 and 2024. The studies investigated RT-induced immune stimulation at optimal HFRT improves AE and clinical outcomes. The systematic search for relevant studies was carried out using the following keywords: RT, hypo-fractionation RT, immune system, anti-tumour CD8+ T, Infiltration of CD8+ cytotoxic T-cells, tumour-specific, monocytic myeloid-derived suppressor cells (M-MDSCs), immune stimulation, RT-schedule, RT-dose, RT-fraction, and clinical outcome. Similarly, we performed a manual review of references to select additional studies. Table 1 summarises the search strategy of this systematic review.
Selection (inclusion and exclusion) criteria
2.2
The titles and abstracts of relevant studies were evaluated for their contents, ensuring adherence to this systematic review article’s inclusion and exclusion criteria. Inclusion criteria were (I) the studies investigating RT-induced immune stimulation; (II) the studies investigating immune cells such as CD8+ cytotoxic T-cells, regulatory T-cells (Tregs), and M-MDSCs after using HFRT; (III) the studies monitoring optimal RT-type, RT-dose, RT-fraction, RT-schedule, (IV) the studies investigating AE after using HFRT, time to AE, and site of AE; (IV) the studies recorded patient’s characteristics and association with the clinical outcome; and (V) the studies analysed the association of HFRT-induced immune stimulation and improved clinical outcomes including complete response (CR), partial response (PR) and stable disease (SD).
The exclusion criteria for this systematic review were (I) editorials, (II) case reports, (III) studies did not have primary data, (IV) studies did not report bystander effect (BE) and AE following HFRT in metastatic disease, (V) studies monitoring RT with Immunotherapy/Chemotherapy combination, (VI) studies which were not written in English, and (VII) did not have full text available. The articles that fulfilled the inclusion criteria were shortlisted, and the primary characteristics are summarised in Table 2.
Data extraction and quality assessment method
2.3
The data were extracted from selected studies by two authors. The extracted data included (I) general information (first author, publication’s year, method of patient recruitment, and sampling methods); (II) clinical characteristics (T-stage, age, treatment option, RT-type, RT-dose, RT-fraction, and RT-schedule); (III) T-cell response following HFRT and (IV) clinical outcomes (time to AE, site of AE, biochemical recurrence, side effects of RT or RT-induced toxicity, treatment response (CR and PR), tumour control, PFS and OS).
The study’s quality was assessed using the PROBAST (Prediction Model Risk of Bias Assessment Tool), which evaluates the applicability and risk of bias in diagnostic tests (30). To address discrepancies in interpretation, two assessors jointly assessed one article first. Articles were then scored for each study, and section deficiencies were noted for further discussion. The relevant published articles were retrieved and imported into an Endnote X21 database (31). Analogous articles were identified and deleted using the Endnote’s duplicate function. We considered studies only describing multivariable-adjusted hazard ratios (aHR). Moreover, we excluded studies that reported crude or unadjusted outcome measures between patients treated with HFRT.
Results
3
Systematic review analysis
3.1
The literature search identified 1431 preclinical and clinical studies: 140 from PubMed, 550 from Science Direct, and 741 from Google Scholar, respectively. Of these 1431 studies, 832 were excluded after reviewing the titles and abstracts, and 599 were selected at the first screening stage. At the second screening stage, 550 studies were removed after full-text examination, and 14 were selected. Furthermore, 49 studies were assessed for eligibility, and 34 studies were removed for the following reasons: (1) case report = 5; (2) editorial = 5; (3) lack of present primary data = 4; (4) lack of bystander and abscopal information = 3; (5) no full text available = 7; (6) studies investigating RT effect in combination with Immunotherapy/Chemotherapy = 13. After eligibility consideration, 12 (7 = preclinical and 5 = clinical) studies were selected. Figure 1 shows our literature search and selection strategy as a flowchart.
Representation of the PRISMA workflow for selecting studies.
T-cell response following HFRT
3.2
To evaluate the potential immunogenic modulation of HFRT at the optimal schedule, 10 of the 12 selected studies have observed T-cell infiltration to the tumour environment following HFRT (Table 3) (5, 32–40).
Out of 10, 7 preclinical studies reported an increased infiltration of T-cells to the tumour microenvironment after HFRT (5, 32–34, 36, 38, 39). For example, Filatenkov et al. reported that the unirradiated tumour with HFRT contained approximately 19% CD8+ T-cells, the irradiated tumour contained approximately 70% at day 35, and the percentage of MDSCs decreased after day 24 (5). In addition, there was a trend toward an increase in CD8+ T-cells in both irradiated and non-irradiated parts of the tumour seven days post-10 Gy RT (32). Kim et al. reported that five-day spacing was more effective than a one-day interval in enhancing anti-tumour immunity via activating the CD8+ T-cells and suppressing the M-MDSCs (33).
Some studies have demonstrated that optimal RT dose and fraction can cause immunologic effects and increased CD8+ T-cell infiltration in the tumour microenvironment (34, 36, 38, 39). Fray et al. stated that on day 8, more cytotoxic T-cells and a lower percentage of Tregs (CD4+/CD25+/FoxP3+) were identified in the irradiated tumours using irradiation two fractions × 5 Gy (34). In addition, an increase in CD8+ T-cells concentration was observed from 48 h to 3 weeks after HFRT in 4.6 Gy × 3 fractions and 6.2 Gy × 2 fractions (p < 0.05), but not in 3.7 Gy × 4 fractions and 10 Gy × 1 fraction (36). A single HFRT dose of 15 Gy increased CD8+ T-cell responses and decreased Tregs (38). The increased proportion of CD8+ T-cells was noticed on day seven after the first HFRT session in the 1 Fraction × 16.4 Gy group (p = 0.002), 3 fractions × 8 Gy group (p < 0.001), and in the 18 fractions × 2 Gy group (p < 0.001); versus 1.4% ± 0.3% in the control group (39).
In the clinical study group, three studies also stated the effect of HFRT on T-cell infiltration in cancer patients (35, 37, 40). Zhang et al. demonstrated that HFRT increased the frequency of CD8+ T-cell infiltration but decreased the frequency of inhibitory Tregs (35). Moreover, Muraro et al. also identified that half of the patients showed increased numbers of activated natural killer (NK) cells and T-cells (CD4+ and CD8+) immediately after the first dose of SBRT (37). Additionally, activated CD25+ CD4+ memory T-cells and CD25+ CD8+ memory T-cells increased following SAR to parenchymal sites, not bone or brain (40).
Tumour control following HFRT
3.3
To evaluate the efficacy of HFRT delivered in various schedules, 6 out of 12 selected preclinical and clinical studies tested the HFRT schemes with several-day intervals to control tumour growth (Table 4) (33, 34, 36, 38, 39, 41).
In the preclinical studies group, 5 out of 6 reported tumour control after HFRT (33, 34, 36, 38, 39). Kim et al. reported that tumour growth delays by a five-day interval RT (p = 0.0293) or a seven-day interval RT (p = 0.0434) were more significant than those by a one-day interval (p = 0.6413) (33). Moreover, tumour growth was significantly delayed in the mice irradiated with 2 fractions × 5 Gy in a 4-day interval (34).
To evaluate the tumour control at different RT schedules, Zhao et al. reported that tumour growth was considerably delayed in the 6.2 Gy × 2 fractions group compared with the control group (p < 0.01) (36). Furthermore, the group receiving local single-dose HFRT at 7.5 and 15 Gy showed significant tumour control, whereas the group receiving 5 Gy had a minimal effect (38). In addition, Grapin et al. monitored the tumour’s growth with 18 fractions × 2 Gy and 3 fractions × 8 Gy regimens and found the most extended tumour growth delay compared to 1 fraction × 16.4 Gy (39).
On the other hand, only one clinical study reported that the bulky tumour control rate was 95% for the SBRT groups compared with 20% in the other two groups (41).
Consequent vaccine-like effect following HFRT
3.4
BE, or AE effect of HFRT, is a rare and unpredictable outcome encountered during the metastatic treatment, where tumour regression is observed to be distant from the irradiated volume. Eight previous preclinical and clinical studies have reported AE and clinical outcomes at optimal HFRT schedules (Table 5) (5, 32, 35–37, 40–42).
In the preclinical studies group, three studies have observed immunological effects and response rates following the use of HFRT. For example, 13 of the 14 mice achieved complete remissions when treated with 30 Gy, while 3 of 5 developed complete tumour remissions when the HFRT dose was specified at 20 Gy (5). Eight 67NR models (35%) experienced a significant AE after partial irradiation with a single dose of 10 Gy (32). Another preclinical study also stated that those treated with 6.2 Gy × 2 fractions showed a noteworthy improvement in OS compared to the control group (36).
In the clinical studies group, Kim et al. observed a better OS in patients treated with HFRT regimens of 48 Gy × 6 fractions or 48 Gy × 8 fractions, which activate the immune system three weeks after treatment (35). The patients showed increased numbers of activated natural killer (NK) cells immediately after the first SBRT dose, showing better PFS (37). Authors from another study have identified an AE in lung and liver cancer patients treated with 1–5 fractions of SBRT or 1–10 fractions of HCRT, but it was not observed in bone and brain (40).
Moreover, Tubin et al. observed AE in 45% (9/20) of patients treated with SBRT (41). They also observed that SBRT was more likely to improve survival OS rates (p = 0.099), cancer-specific survival (CSS) (p = 0.049) and PFS rates (p = 0.003) (41). Another Tubin study reported significant BE and AE response rates of 96% (22/23 patients) and 52% (12/23 patients), and improved OS 70% (16/23) and PFS 87% (20/23) rates, respectively, in patients whose bulky tumours were partially irradiated (42).
Discussion
4
Though RT has long been used in cancer therapy, it has a history of immunosuppressive side effects. Researchers believe that lymphopenia can result from localised RT, which includes radiation to the chest or central nervous system (43, 44). The leading causes of this are the radiation exposure of the bloodstream and the inherent radiation sensitivity of immune cells, even at low radiation doses (<1 Gy) (43–45). Although radiation has long been believed to suppress the immune system, there is a bunch of evidence showing that radiation can, under certain conditions, actually increase the immune system’s ability to fight cancer (5, 27, 32–41, 46–48).
Established tumour cells often lose their capacity to present antigens through various genetic and epigenetic mechanisms, enabling them to avoid the immune system. Radiation’s direct cytotoxic effects may cause the release of tumour-specific antigens, which can then prompt antigen-presenting cells to trigger a T-cell immune response (49). Although dendritic cells can present tumour antigens to T cells, the successful activation of tumour antigen-specific T-cell immunity requires additional danger signals to enhance T-cell activation (49). Therefore, during radiation-induced cell death, both tumour antigen release and presentation are improved, helping to activate an immune response (50). These specific events following radiation-induced tumour cell killing have led to the concept of utilising RT as a method of in situ vaccination” (51, 52).
Considering the increasing evidence that underlying anti-immune responses may be essential in eradicating certain tumours with SBRT, investigations have been conducted to delineate optimal radiation schedules for maximising anti-tumoural immunity in animal models (53, 54). Marciscano et al. extensively reviewed past studies on the optimal dose and fractionation schedule for increasing anti-tumoural immunity (55). Bae et al. reported that three days of fraction intervals significantly decreased gastrointestinal complications without impairing the tumour control rate of SABR in hepatocellular carcinoma (14). Moreover, using immunological hot and cold tumours, researchers also compared anti-tumoural immunity exposed to two fractions of irradiation administered on consecutive days or at intervals of 5 days in the mouse model (33).
Furthermore, when radiation is administered at moderate or higher dose fractions, local RT can activate CD8+ cytotoxic T-cells involved in both local and systemic tumour control (abscopal) (24, 46, 56). Therefore, in previous studies, RT with 3 to 5 doses of <10 to 12 Gy appears particularly immunogenic (11, 38, 57–59). Some earlier studies revealed that hRT elevates CD8+ concentration between days 5 and 8 after hRT (34, 60, 61). Filatenkov et al. reported that irradiation with 1 Fraction × 30 Gy was curative and induced protective CD8+ T-cell-mediated immunity (5). A similar protracted schedule (4 fractions × 5 Gy over 14 days) failed to locally control B16 melanoma tumours expressing a model antigen with a low total dose of RT and large inter-fraction intervals; however, a single 20 Gy fraction did so (46). Moreover, SRS with a single dose of at least 30 Gy has been suggested to be more effective than daily fractionated radiation (2, 6).
Several researchers have previously reported substantial immune effects and tumour reduction/cure through selective and time-dependent RT, which targets the immune system instead of the tumour (62–64). The effectiveness of these methods depended on the ability to determine when Tregs were dividing synchronously and periodically during cell division (65). At this brief window in time (mitosis), the Tregs were highly sensitive to selective ablation, thus mitigating or removing their homeostatic immunosuppressive effects on other tumour-specific immune cells not in mitosis at that specific time point (64). Due to the tumour’s underlying immunology, RT may evolve towards more “immunologically relevant” schedules to break tumour tolerance locally and systemically (66, 67).
Contrary to the results of RT studies, some studies, in combination with immunotherapy, found no evidence of AE and response rate after using HFRT (68, 69). For example, McBride’s and Kim et al. studies found no evidence of AE and improved clinical outcomes by adding SBRT to nivolumab and Nivolumab plus ipilimumab in patients with metastatic head and neck squamous cell carcinoma (HNSCC) and Advanced Merkel Cell Carcinoma, respectively (68, 69). The small sample size may have contributed to the lack of evidence of an additional benefit or support for AE with the addition of SBRT, as mentioned in these clinical trials. Some previous studies have shown potential therapeutic benefits with systemic therapies given at the right time to selectively ablate synchronously dividing suppressor T cells (now called Regulatory T Cells) while sparing the effector T cells (63, 70, 71). Therefore, it suggests that the timing of immunotherapy and RT may play a role in treatment efficacy via immune modulation. We believe that additional investigation is warranted to determine the optimal RT dose and timing, immunotherapeutic agent, and large patient cohort to fully evaluate the potential of the AE on the response rate.
Conclusions
5
Our systematic review data revealed that HFRT with an optimal regimen can induce the activation of T lymphocytes while simultaneously reducing the frequency of Tregs. These studies also suggested that optimal dosages and fractions of HFRT induce immune response. However, it should be further explored to provide clinicians with information that would be valuable when making decisions regarding patient care. This strategy may increase cancer patients’ response rate to treatment, lower the cost and length of treatment and lower thir chance of developing immune-related side effects and general toxicity after receiving chemotherapy and immunotherapy.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Durante M Reppingen N Held KD. Immunologically augmented cancer treatment using modern radiotherapy. Trends Mol Med. (2013) 19:565–82. doi: 10.1016/j.molmed.2013.05.007, PMID: 23831337 · doi ↗ · pubmed ↗
- 2Goodman KA Wiegner EA Maturen KE Zhang Z Mo Q Yang G. Dose-escalation study of single-fraction stereotactic body radiotherapy for liver Malignancies. Int J Radiat oncology biology physics. (2010) 78:486–93. doi: 10.1016/j.ijrobp.2009.08.020, PMID: 20350791 · doi ↗ · pubmed ↗
- 3Favaudon V Caplier L Monceau V Pouzoulet F Sayarath M Fouillade C. Ultrahigh dose-rate FLASH irradiation increases the differential response between normal and tumor tissue in mice. Sci Trans Med. (2014) 6:245ra 93. doi: 10.1126/scitranslmed.3008973, PMID: 25031268 · doi ↗ · pubmed ↗
- 4Park C Papiez L Zhang S Story M Timmerman RD. Universal survival curve and single fraction equivalent dose: useful tools in understanding potency of ablative radiotherapy. Int J Radiat oncology biology physics. (2008) 70:847–52. doi: 10.1016/j.ijrobp.2007.10.059, PMID: 18262098 · doi ↗ · pubmed ↗
- 5Filatenkov A Baker J Mueller AM Kenkel J Ahn GO Dutt S. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin Cancer Res. (2015) 21:3727–39. doi: 10.1158/1078-0432.CCR-14-2824, PMID: 25869387 PMC 4537844 · doi ↗ · pubmed ↗
- 6Loo BW Jr. Stereotactic ablative radiotherapy (SABR) for lung cancer: What does the future hold? J Thorac Dis. (2011) 3:150–2. doi: 10.3978/j.issn.2072-1439.2011.06.08, PMID: 22263081 PMC 3256516 · doi ↗ · pubmed ↗
- 7Jaffray DA. Image-guided radiotherapy: from current concept to future perspectives. Nat Rev Clin Oncol. (2012) 9:688–99. doi: 10.1038/nrclinonc.2012.194, PMID: 23165124 · doi ↗ · pubmed ↗
- 8Bae SH Kim MS Jang WI Kay CS Kim W Kim ES. A survey of stereotactic body radiotherapy in Korea. Cancer Res Treat. (2015) 47:379–86. doi: 10.4143/crt.2014.021, PMID: 25578057 PMC 4506107 · doi ↗ · pubmed ↗