GM2 Gangliosidosis AB Variant: A Hidden Truth
Inês Noites, Ana Sofia Coelho, Catarina Magalhães, Sandra Ramos, Francisco Laranjeira, Lúcia Lacerda, Ricardo Taipa, Cristina Garrido, Teresa Temudo, Sónia Figueiroa

TL;DR
This paper reports the first case of GM2AB in a Portuguese patient, emphasizing the importance of genetic testing for rare neurodegenerative disorders.
Contribution
The paper presents the first documented case of GM2AB in Portugal and highlights the role of post-mortem genetic analysis in diagnosing rare diseases.
Findings
A Portuguese patient with GM2AB was diagnosed through post-mortem genetic sequencing.
The patient had a homozygous pathogenic mutation in the GM2A gene.
The case underscores the need to consider GM2AB in neurodegenerative disorders with normal enzyme activity.
Abstract
GM2 gangliosidosis AB variant (GM2AB) is a rare neurodegenerative lysosomal storage disorder with clinical features resembling Tay-Sachs disease but characterized by normal lysosomal β-hexosaminidase A enzyme activity. To date, only 14 cases of the acute infantile form have been reported. To the best of our knowledge, this is the first case of GM2AB in a Portuguese patient reported in the literature. We describe the case of a girl with GM2AB, whose clinical presentation and pathological findings were critical for diagnosis. Post-mortem genetic sequencing identified a pathogenic mutation in homozygosity in the GM2A gene, confirming the diagnosis. This case highlights the importance of considering GM2AB in patients with severe neurodegenerative phenotypes and typical pathological findings, even when enzymatic studies are normal. Preserving genetic material post-mortem may allow for…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
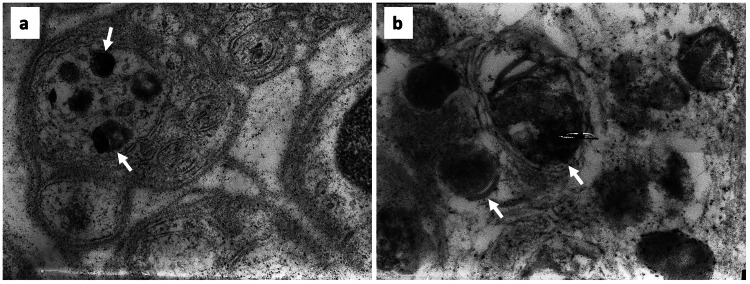 Figure 1
Figure 1| Analytical parameters | Value | Reference interval |
| Lactate | 1.8 | 0.50-2.20 mmol/L |
| Pyruvate | 85.7 | 34-103 μmol/L |
| Ammonia | 37.5 | 26-47 μmol/L |
| Acylcarnitine profile | Within normal limits* | - |
| Plasma amino acid chromatography | Within normal limits* | - |
| Urine amino acid chromatography | Within normal limits* | - |
| β-Galactosidase | 322 | 73.0-585 nmol/h/mg protein |
| β-Hexosaminidase A | 250 | 185-962 nmol/h/mg protein |
| Total β-hexosaminidase | 4798 | 3593-30182 nmol/h/mg protein |
| Galactocerebrosidase | 2.6 | 0.71-3.59 nmol/h/mg protein |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLysosomal Storage Disorders Research · Cellular transport and secretion · Erythrocyte Function and Pathophysiology
Introduction
GM2 gangliosidoses are a group of rare autosomal recessive lysosomal storage disorders characterized by the pathological accumulation of GM2 gangliosides in the central nervous system (CNS) [1]. These intra-lysosomal deposits lead to progressive cell death, demyelination, and neurodegeneration [2].
The GM2 gangliosidosis AB variant (GM2AB) is distinguished among these disorders by its unique genetic and biochemical characteristics. While Tay-Sachs and Sandhoff diseases are caused by mutations in the HEXA and HEXB genes, respectively, GM2AB results from mutations in the GM2A gene [1,3]. This gene encodes the GM2 activator protein, a non-catalytic cofactor required for the hydrolysis of GM2 gangliosides by the β-hexosaminidase A enzyme complex [2].
A characteristic feature of GM2AB, shared with other GM2 gangliosidoses, is the presence of a cherry-red spot in the retina [1,2,4-9]. This distinctive finding results from ganglioside accumulation in the retinal ganglion cells [1,9], causing the retina to appear whitish and degenerated. In contrast, the macula, devoid of ganglion cells, appears reddish against the pale surrounding retina. This particular feature serves as a crucial diagnostic marker in the clinical evaluation of suspected GM2 gangliosidosis variants [3,4], suggesting that an ophthalmological examination could be useful in the diagnostic workup.
Clinically, GM2AB manifests with a spectrum of symptoms consistent with those observed in Tay-Sachs and Sandhoff diseases [2-4,8]. The acute infantile form of this condition typically presents between four and 12 months of age [5]. Early signs include developmental delay [2-5,8], muscle weakness [1,4,5,8], and exaggerated startle response to auditory stimuli [2,4-6,8]. As the disease progresses, severe neurological deterioration occurs, manifesting as seizures, vision and hearing loss, intellectual disability, and motor dysfunction [4,5,8]. Affected individuals often succumb to the disease within the first few years of life, most commonly from respiratory complications such as recurrent infections and aspiration, secondary to progressive neurological deterioration [5]. The rate of progression and phenotypic severity may vary depending on the predicted impact of the specific mutation on the function of the GM2 activator protein [6,7]. Currently, there is no cure for GM2AB, and treatment focuses on supportive measures to manage symptoms [5].
Pathologically, GM2AB is characterized by significant neurodegeneration within the CNS because of GM2 ganglioside accumulation [1,2,5]. Histological examination reveals widespread neuronal storage, with neurons appearing distended and filled with membranous cytoplasmic bodies [3]. These structures are most prominent in the cerebral cortex, cerebellum, brainstem, and spinal cord. Electron microscopy often reveals multilamellar membrane structures within lysosomes, known as zebra bodies, indicative of lysosomal dysfunction and ganglioside storage [9].
Although the CNS is primarily affected, other tissues, including skin, may also exhibit evidence of ganglioside storage, reflecting the disorder’s multi-systemic nature [5,9]. In GM2AB, routine biochemical assays, including β-hexosaminidase A and B activity, are typically normal, in contrast to Tay-Sachs and Sandhoff disease [4,5]. There are no routine biochemical markers specific for GM2AB. Therefore, diagnosis relies primarily on genetic testing of the GM2A gene, which may include single-gene analysis, multigene panels, or comprehensive approaches such as exome sequencing, depending on the clinical context. Specialized functional assays for GM2 activator protein deficiency are available in research settings [2,5,7].
Currently, only 14 cases of the acute infantile presentation of GM2AB have been reported in the literature, highlighting the rarity of this disorder [1,5].
Case presentation
We present the case of a girl born to healthy, non-consanguineous Portuguese parents. She was delivered via cesarean section at term, with Apgar scores of 6 at one minute and 10 at five minutes. The infant’s early neonatal course was unremarkable, with a normal physical examination and uneventful discharge.
During the first year of life, the patient achieved developmental milestones as expected for age: rolling by four months, sitting unsupported by six months, developing a pincer grasp by nine months, and first words by 11 months. The head circumference was around the 50th percentile at birth, then decreased to below the 10th percentile by five months, preceding subsequent developmental regression. Between 12 and 17 months of age, she experienced a progressive loss of psychomotor skills, initially presenting with cerebellar ataxia and an inability to walk independently. Over time, she lost the ability to sit without support and exhibited choreoathetotic movements of her upper limbs. Regression also affected speech, social interaction, and feeding abilities. By 14 months, she had lost visual attentiveness and began to suffer from generalized tonic-clonic and myoclonic seizures.
Physical examination revealed several notable findings, including microcephaly, non-dysmorphic facial features, episodic deviations of the upper gaze, and a pronounced acoustic startle reflex. Additionally, the child exhibited profound axial hypotonia, pyramidal signs, and a bilateral cherry-red spot on ophthalmologic examination.
Brain MRI findings in the GM2AB variant are highly variable and do not follow a consistent or specific neuroimaging pattern. While some affected infants have been reported with normal brain MRI [1,5,8], others demonstrate abnormal T2 signal intensities in the periventricular white matter and basal ganglia [2,8]. Delayed myelination and T2 hyperintensities in subcortical white matter, basal ganglia, and thalami have also been described [5]. In our patient, the presence of periventricular white matter hyperintensities is in line with previously reported abnormalities, although the overall spectrum of MRI changes in GM2AB remains heterogeneous and nonspecific. A comprehensive laboratory workup (Table 1) showed normal liver and kidney function tests and normal lactate, pyruvate, and ammonia levels. Plasma and urine amino acid chromatography, organic acid excretion, and acylcarnitine profile analysis were all within normal limits. Urinary sulfatides and glycosaminoglycans were also in the normal range. Enzymatic studies conducted on fibroblasts, including assays for β-galactosidase, β-hexosaminidase A and total β-hexosaminidase, sphingomyelinase, galactocerebrosidase, α-neuraminidase, palmitoyl protein thioesterase 1, and tripeptidyl peptidase I, showed normal activity.
Skin biopsy analysis, obtained using the routine technique, provided further diagnostic insights. Electron microscopy revealed intraneuronal lamellar inclusions (Figure 1), and toluidine blue staining confirmed the presence of neuronal storage material, consistent with a lysosomal storage disorder. Despite normal enzymatic assay results, these ultrastructural features substantially reinforced the clinical suspicion.
Electron microscopy of skin biopsy showing electron-dense inclusions (arrow) in the cytoplasm of non-myelinated axons. These inclusions are amorphous but occasionally exhibit a lamellar structure at the periphery (a. 20,000x magnification; b. 40,000x magnification).
By 35 months of age, her condition had continued to deteriorate. She became unresponsive to environmental stimuli, lacked visual contact and language skills, exhibited flaccid tetraparesis, and responded only to painful stimuli, ultimately succumbing to pneumonia. Despite extensive diagnostic efforts, including comprehensive laboratory and enzymatic studies, a definitive etiology remained unidentified during her lifetime.
Years later, with the advent of GM2A gene sequencing at our institution, a homozygous c.333delC p.(Cys112Valfs*7) mutation was identified, confirming the diagnosis of GM2AB. This frameshift variant causes the change of 6 amino acids after position 111 before reaching a premature stop codon. As a result, a truncated protein is produced, approximately in the middle of its natural length, which loses all bisulfide bonds and affects its three-dimensional structure. It is also worth mentioning the presence of several heterozygous polymorphic variants spread across the GM2A gene, which corroborate the non-consanguinity of the parents.
At that time, the diagnosis was also confirmed through next-generation sequencing, as the case was included in a research project [10] that aimed to evaluate the use of this methodology for genetic diagnosis.
Discussion
GM2 gangliosidoses are a group of rare and complex lysosomal storage disorders characterized by intricate clinical, biochemical, and molecular features [1,3,7]. These conditions result from mutations in genes that regulate the breakdown of GM2 ganglioside [2,3], a critical lipid component of neuronal membranes. The degradation of GM2 involves several enzymatic steps [3], with the disruption of this process leading to the accumulation of GM2 gangliosides [2], primarily in the CNS, which causes severe neurodegeneration [1,2,5].
The pathophysiology of GM2 gangliosidosis is closely linked to ganglioside metabolism in neuronal cells, resulting in the CNS being the main site of disease. Therefore, there is a characteristic clinical presentation across the different forms of GM2 gangliosidosis, marked by progressive neurological deterioration [1,4]. Key diagnostic features, such as the presence of a cherry-red spot on retinal examination and early-onset neurodegenerative symptoms, strongly suggest a lysosomal storage disorder [1,4-6,8].
This is the first documented case of this rare variant in a Portuguese patient reported in the literature. In this patient, the normal enzymatic activity of β-hexosaminidases A and B initially masked the diagnosis. At that time, genetic testing for the GM2A gene was not available, so a biopsy was performed, which provided supportive pathological findings. The patient’s clinical presentation and pathological findings provided essential clues, prompting further investigation. Currently, when enzyme results are normal despite high clinical suspicion, ophthalmologic evaluation and confirmatory genetic testing should be prioritized.
Ultimately, genetic sequencing conducted years after the patient’s death identified a homozygous c.333delC p.(Cys112Valfs*7) mutation in the GM2A gene, confirming the diagnosis of GM2AB. To date, most GM2A mutations associated with GM2AB gangliosidosis are missense variants, which are thought to disrupt the GM2 activator protein by impairing its stability or lipid-binding capacity [2,8,9]. The homozygous frameshift mutation identified in this patient leads to a premature stop codon and truncation of the protein, severely compromising its three-dimensional structure. Truncating variants, which include both frameshift and rare nonsense mutations, are generally predicted to result in a more severe loss of function than missense substitutions. Given the small number of reported cases, no clear genotype-phenotype correlation has yet been established [5].
Genetic confirmation clarified the patient’s clinical course and emphasized the pivotal role of molecular diagnostics in identifying complex neurodegenerative disorders and guiding management strategies for affected families.
The diagnostic challenges faced in this case highlight the importance of considering GM2 gangliosidosis in the differential diagnosis, even when conventional enzymatic tests return normal results. The diagnosis of GM2AB, established through gene sequencing, underscores the need for molecular testing in atypical or unresolved cases.
Conclusions
Currently, GM2 gangliosidosis remains an incurable neurodegenerative disorder with high morbidity and mortality. Nevertheless, an accurate diagnosis is essential for providing optimal supportive care and genetic counselling to affected families. While early recognition of the disease can facilitate appropriate management strategies, the lack of effective therapies emphasizes the need for continued research into potential treatments. This case highlights the critical importance of retaining genetic material from patients with atypical or unresolved cases, particularly those with severe phenotypes. As demonstrated here, advancements in genetic technology can enable definitive diagnoses, even years after a patient’s death. Such diagnoses provide closure for families, inform genetic counseling, and guide decisions regarding future pregnancies, underscoring both the medical and psychosocial value of molecular testing.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Multimodal optical imaging and genetic features of AB variant GM 2 gangliosidosis: a case report Front Pediatr Chen Q Lu F 11478361120233721558910.3389/fped.2023.1147836 PMC 10196110 · doi ↗ · pubmed ↗
- 2GM 2 gangliosidosis AB variant: novel mutation from India - a case report with a review BMC Pediatr Sheth J Datar C Mistri M Bhavsar R Sheth F Shah K 881620162740209110.1186/s 12887-016-0626-6PMC 4939586 · doi ↗ · pubmed ↗
- 3GM 2 activator deficiency caused by a homozygous exon 2 deletion in GM 2AJIMD Rep Hall PL Laine R Alexander JJ Ankala A Teot LA Lidov HG Anselm I 61653820182854063610.1007/8904_2017_31PMC 5874204 · doi ↗ · pubmed ↗
- 4Two patients from Turkey with a novel variant in the GM 2A gene and review of the literature J Pediatr Endocrinol Metab İnci A Cengiz Ergin FB Biberoğlu G Okurİ EzgüFS Tümer L 8058123420213381941510.1515/jpem-2020-0655 · doi ↗ · pubmed ↗
- 5GM 2 activator deficiency Gene Reviews® [Internet] Xiao C Toro C Tifft C Seattle (WA)University of Washington 2022 https://www.ncbi.nlm.nih.gov/books/NBK 583219/36007105 · pubmed ↗
- 6Mutation in GM 2A leads to a progressive chorea-dementia syndrome Tremor Other Hyperkinet Mov (N Y) Salih MA Seidahmed MZ El Khashab HY 306520152620340210.7916/D 8D 21WQ 0PMC 4502426 · doi ↗ · pubmed ↗
- 7Atypical juvenile presentation of GM 2 gangliosidosis AB in a patient compound-heterozygote for c.259G > T and c.164C > T mutations in the GM 2A gene Mol Genet Metab Rep Martins C Brunel-Guitton C Lortie A Gauvin F Morales CR Mitchell GA Pshezhetsky AV 24291120172841707210.1016/j.ymgmr.2017.01.017PMC 5388932 · doi ↗ · pubmed ↗
- 8GM 2-gangliosidosis, AB variant: clinical, ophthalmological, MRI, and molecular findings JIMD Rep Renaud D Brodsky M 83862520162608232710.1007/8904_2015_469PMC 5059184 · doi ↗ · pubmed ↗