A Systematic Review on the Role of the Stria Vascularis in Menière’s Disease Pathogenesis
Pablo Cruz-Granados, Sreeparna Das, Kiana Bagheri-Loftabad, Jose A. Lopez-Escamez

TL;DR
This review explores how genes in the stria vascularis may contribute to Menière’s Disease, focusing on immune and auditory pathways.
Contribution
A systematic identification of SV cell-type-specific genes and their potential roles in Menière’s Disease pathogenesis.
Findings
Seven immune-related and six auditory-related genes were identified in SV cell types.
Gene-set-enrichment analysis linked SV genes to gap-junction assembly and immune pathways.
TWEAK signaling between SV cell types may drive inflammation in Menière’s Disease.
Abstract
The stria vascularis (SV) is a secretory epithelium that maintains fluid homeostasis and generates the endocochlear potential in the cochlear duct. Multiomic studies have identified genes in the SV that could contribute to the pathogenesis of Menière’s Disease (MD), a disorder defined by episodic vertigo, sensorineural hearing loss, and tinnitus. This systematic review identified genes expressed in the SV cell types (marginal, intermediate, and basal) and gap junction proteins to evaluate their pathophysiological connections to MD. We conducted a literature search on 1293 articles relevant to MD and SV that were screened for SV genes involved in MD. Following quality assessment, 130 studies met the inclusion criteria, comprising 26 human studies, 101 animal studies, and three human-animal studies. Seven immune-related and six auditory-related genes were identified: CACNA1D, ESRRB,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
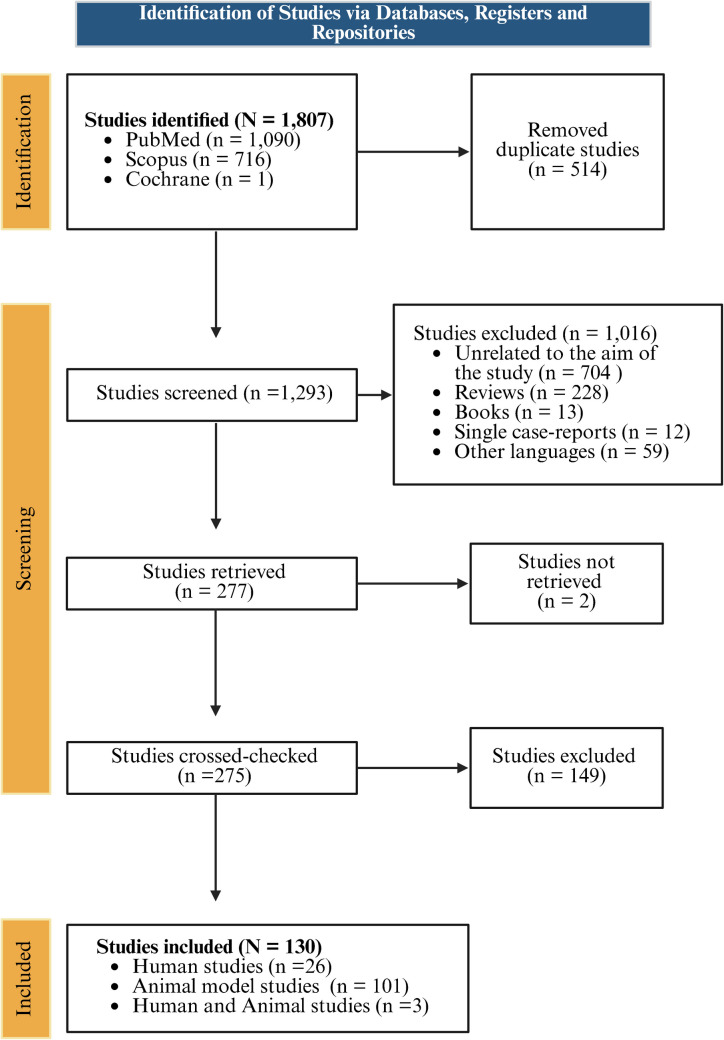 Figure 1
Figure 1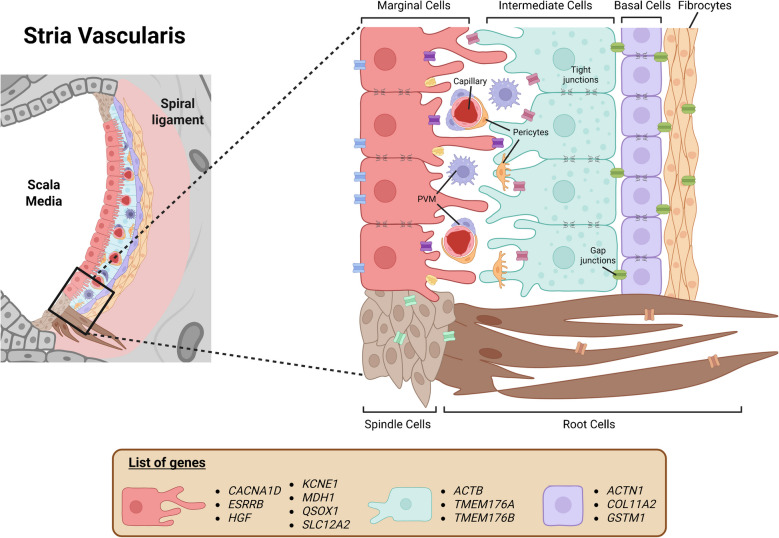 Figure 2
Figure 2- —http://dx.doi.org/10.13039/501100001774University of Sydney
- —The University of Sydney
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHearing, Cochlea, Tinnitus, Genetics · Vestibular and auditory disorders · Connexins and lens biology
Introduction
Menière’s Disease (MD) is an inner ear syndrome characterized by sensorineural hearing loss (SNHL) with tinnitus and episodes of vertigo of multifactorial origin, with a significant heritability according to familial aggregation and sequencing studies in familial and sporadic cases [1]. Menière’s Disease is prevalent across Europe and East Asia, with a greater number of cases reported in the European population [2]. Several MD clinical subgroups have been described according to the familial history of MD and associated comorbidities, including migraine and autoimmune conditions [3]. Additionally, distinct phenotypes have been identified, according to their cytokine profiles and systemic inflammation, with one group exhibiting higher levels of Th2 cytokines, IgE and a type 2 immune response, and another one with high levels of IL-1β and autoinflammation [4–6].
The accumulation of endolymph fluid in the scala media is associated with the pathophysiology of MD. The endolymphatic hydrops (EH) leads to increased pressure within the cochlear duct with an enlargement of the endolymphatic space, which results in damage to the organ of Corti and other inner ear membranes. However, vertigo attacks, hearing loss, or tinnitus in MD are not explained by EH alone, as individuals without MD have been found with EH [7].
The stria vascularis (SV) is an organ made up of three main cell layers, located in the lateral wall of the cochlear duct [8]. It is surrounded by the spiral ligament and plays a crucial role in regulating K^+^ homeostasis in the scala media and generating the endocochlear potential, which is essential for hearing [9, 10].
Exome sequencing and genome sequencing in SNHL and MD have identified a significant number of genes with a burden of rare variation in differentially expressed genes (DEG) in the SV [11, 12].
This review aims to understand the importance and role of SV genes in the development of MD.
Methodology
This review was conducted following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) [13] guidelines for systematic reviews, and it abided by the Meta-analyses Of Observational Studies in Epidemiology (MOOSE) [14] checklist. This review’s protocol was registered on PROSPERO (CRD42024618482).
The PICO (Participants–Intervention–Comparison–Outcomes) question included the following:
- Participants: studies including datasets from individuals with definite Sporadic MD or Familial MD, or MD animal models, or cellular models.
- Intervention: epigenomic, genomic, transcriptomic, or proteomic studies related to the stria vascularis and genes linked to MD.
- Comparison: healthy controls, individuals without MD, or animal models not affected by the disease.
- Main outcome: identification of differentially expressed genes, epigenetic modifications, differentially expressed transcripts, or proteins in the stria vascularis associated with MD onset or progression.
- Secondary outcomes: predicted enriched biological pathways, biomarkers, or mechanisms linked to MD development, particularly relating to endolymphatic hydrops, fluid balance, or inner ear function.
- Study design: case–control studies, cohort studies, animal models, genomic and proteomic analyses of tissue samples, or epigenomic profiling of DNA.
Search Strategy
The search strategy was carried out on 18th November 2024 using original articles published in the year 2005 onwards from PubMed, Scopus, and Cochrane databases. The MeSH terms used were as follows: (Meniere OR Stria Vascularis) AND (Epigenomics OR Epigenetics OR Genomics OR Genetics OR Gene or Transcriptomics OR RNAseq OR Protein). Duplicate articles and articles not relevant to the review’s objective were excluded. Furthermore, the following exclusion criteria were also used:
- Studies published in other languages other than English
- Single-case reports.
Data Collection
Two independent reviewers (P.C.G. and S.D.) reviewed all the abstracts to select records according to the inclusion criteria. When consensus could not be reached, a third reviewer (J.A.L.E.) was involved in resolving discrepancies. We summarized the studies that involved epigenetic changes, genes, transcripts, and proteins from different studies on MD and SV.
Gene Search Using Published Datasets
Further filtering was performed on selected studies to identify those involving genes of interest. DOIs were retrieved, and three independent reviewers (P.C.G., S.D., and K.B.L.) screened the introduction, results, and discussion sections of each study using a tailored list composed of genes associated with Familial MD (FAM136A, DTNA, PRKCB, COCH, DPT, SEMA3D, TECTA, GUSB, SLC6A7, HMX2, LSAMP, OTOG, STRC, MYO7A, and GJD3) [15, 16], marker genes for SV marginal, basal, and intermediate cells from two studies using postnatal day 30 (P30) mice [17, 18] and gap junction proteins (connexins). For SV cell markers, only genes with a ± onefold change (logFC) and > 0.05 p-value adjusted were used. The list of tailored genes can be found in Supplementary Table 1. For genes, the Human Genome Organization (HUGO) nomenclature was used, and for proteins only the recommended name was employed. Both gene and protein identifiers were extracted from UniProt [19] to use as search keywords. Studies that replicated epigenetic changes, genes, transcripts, and/or proteins matching the tailored list were included. Moreover, we retrieved relevant articles to MD and SV by inspecting the reference section of selected papers.
Data Synthesis
Reference information, participant ancestry, study design, sample size, and participants’ age were extracted from studies that reported the selected genes or proteins in the “Results” section.
Risk of Bias Assessment
ROBINS-E and SYRCLE tools [20, 21] were used to assess the risk of bias in non-randomized human and animal studies, respectively. A table was used to summarize the results using a color-coded scale.
Only six ROBINS-E domains were used: (1) confounding-induced bias, (2) bias in exposure measurement, (3) bias in participant selection for the study, (5) bias due to missing data, (6) bias in outcome measurement, and (7) bias in the selection of reported results. Domain (4) bias resulting from post-exposure interventions was excluded as it was irrelevant to this study. For animal and cellular models, items 3, 4, and 5 from SYRCLE were irrelevant and excluded from the analysis.
For studies where both human and animal were used, a mix of ROBINS-E and SYRCLE’s criteria was used.
Hearing and Vestibular Phenotype in Knockout Mice of Stria Vascularis Genes
To explore the role of SV selected genes, auditory brainstem response and vestibular phenotype data were retrieved from the International Mouse Phenotyping Consortium (IMPC) portal for the selected genes in the existing mouse knockout database [22].
MD Gene Expression in the Stria Vascularis
Gene expression data were retrieved from gEAR portal [23] from SV dataset [24] for all genes associated with Familial MD (FAM136A, DTNA, PRKCB, COCH, DPT, SEMA3D, TECTA, GUSB, SLC6A7, HMX2, LSAMP, OTOG, STRC, MYO7A, and GJD3) [15, 16]. A ± 1 logFC filter was used.
Gene Set Enrichment Analysis
GeneCodis v4 [25] was used to perform Gene Set Enrichment Analysis (GSEA) using the overlapped genes between MD and SV employing functional annotations Gene Ontology (GO) Biological Process (BP), GO Cellular Component (CC), GO Molecular Function (MF) and KEGG Pathways, and regulatory annotation CollecTRI TFs.
Results
We selected 130 studies that fitted the inclusion criteria (Fig. 1).1 Of the total number of records, 26 were conducted on humans, and 101 were conducted on animals. Eighty were performed in mice (Mus musculus), 13 in rats (Rattus norvegicus domestica), five in guinea pigs (Cavia spp.), one in flies (Drosophila melanogaster), one in birds (Gallus gallus domesticus and Tyto alba guttata), one in frogs (Xenopus laevis), and one in gerbils (Meriones unguiculatus). Furthermore, three studies contained both human and animal subjects, one study used rats and mice, and one study used murine subjects and frogs (X. laevis).Fig. 1. Flow diagram to select scientific articles from databases and registers for the systematic review
Human Studies
Out of the 26 articles conducted on humans, 15 had genes related to the SV (Table 1) [11, 26–39]. Seven studies were performed on MD, four studies including one on severe tinnitus, one on hereditary SNHL, one on endolymphatic sac, and one on hearing loss, and three did not explore any disease. Three of the studies were cross-sectional, and one was a retrospective, case–control study in silico analysis. None of the studies reported specific gene mutations; instead, they focused on mapping gene expression within the cochlea. These studies identified five genes on SV marginal cells—HSPA4L, KCNE1, SLC12A2, PAX2, and HGF; four on SV intermediate cells—MYO5A, ACTB, KNCJ10, and EYA4; nine genes on SV basal cells—ACTN1, QSOX1, MDH1, COL11A2, ATP1A2, ATP1B3, SLC2A, CLDN11, and ABLIM3; and four connexins—GJB2, GJB6, GJA1, and GJE1. Furthermore, NRCAM, SORBS2, CACNA1D, ESRRB, ATP1B1, TYR, and LMX1A were found to be markers for one or more SV cell types in the tailored list. Two studies also described three Familial MD genes: OTOG, FAM136A, and PRKCB. Table 1. Summary of selected papers reporting genes in human subjects for Meniere disease (MD)DOIAuthor/YearCountryDiseaseDesignSample SizeSex/Median AgeMain ObjectiveGene/Protein—List10.1016/j.ebiom.2021.103309Amanat, S. et al. (2021)SpainSevere tinnitusExome-based phenotype study222NATo identify rare variants in synaptic genes in patients with severe tinnitus using whole-exome sequencingNRCAM—Marginal/intermediate/basal cellsHSPA4LL—Marginal cellsMYO5A—Intermediate cells10.1097/ONO.0000000000000027Arambula, A. et al. (2023)USAMDCase–control; in silico analysis; retrospectiveNANATo determine whether MD enriched perilymph proteins can be localized to specific cochlear cell types using single-cell and single-nucleus RNA-sequencing datasetsNRCAM—Marginal/intermediate/basal cellsACTB—Intermediate cellsACTN1—Basal cellsQSOX1; MDH1—Marginal cells10.1002/jcp.22737Chiarella, G. et al. (2012)ItalyMDProteomic study20Both/51 years oldTo identify the possible biomarkers of MD using a proteomics-driven approachACTB—Intermediate cells10.5152/iao.2019.5076Dai, Q. et al. (2019)ChinaMDComparative study24Both/30–43 years oldTo investigate the gene polymorphisms in Kcne1 and Kcne3 in Familial and Sporadic MD patients compared to healthy controls in a Chinese cohortKCNE1—Marginal cells10.1159/000089410Doi, K. et al. (2005)JapanMDGenetic association study505Both/36–43 years oldTo investigate whether specific single SNPs in the potassium channel genes Kcne1 and Kcne3 are linked to increased susceptibility in MDKCNE1—Marginal cells10.1186/s12864-024–10552-3Fisch, KM. et al. (2024)USAMDCross-sectional study1200NATo identify the rare and common genetic variants associated with unilateral MD using whole genome sequencingSORBS2—Marginal/intermediate cellsCACNA1D—Marginal/intermediate cellsCOL11A2—Basal cellsOTOG—Familial MD10.3389/fgene.2019.00076Gallego-Martinez, A. et al. (2019)SpainMDTargeted-sequence study890Both/NATo assess the burden of rare variants of SNHL genes in Sporadic MDESRRB—Marginal/intermediate cellsFAM136A—Familial MDPRKCB—Familial MDGJB2—Connexins10.1007/s00441-010–0975-7Ishiyama, G. et al. (2010)USAMD/Acoustic neuromaExperimental study18Both/49–87 years oldTo investigate the localization and expressions of aquaporins (Aqp1, Aqp4 and Aqp6) and proteins involved in endolymphatic homeostasis (Na⁺K⁺ATPase and Nkcc1) in MD patientsSLC12A2—Marginal cells10.3389/fnmol.2022.973646Liu, W. & Rask-Andersen, H. (2022)SwedenNACross-sectional study4NATo characterize the distribution of Gjb2 and Gjb6 gene transcripts in distinct cell types of the adult human cochlea using RNAscope® in situ hybridizationKCNJ10—Intermediate cellsCLDN11—Basal cellsATP1A2—Basal cellsGJB2—ConnexinsGJB6—Connexins10.3389/fnmol.2022.857216Liu, W. & Rask-Andersen, H. (2022)SwedenNACross-sectional study4NATo map the expression and cellular distribution of Atp1a1, Atp1b1, and Atp1a3 gene transcripts encoding Na/K-ATPase isoforms in the adult human cochlea using RNAscope® in situ hybridizationATP1B1—Marginal/intermediate/basal cellsATP1B3—Basal cellsGJB2—ConnexinsGJB6—Connexins10.1002/dneu.22279Locher, H. et al. (2015)The Netherlands; BelgiumHereditary SNHLCross-sectional study9NA/W9-18To investigate the embryological development of the human fetal stria vascularis between 9 and 18 weeks of gestation to characterize the expression key potassium-regulating and gap junction proteins involved in hereditary sensorineural hearing loss (SNHL)KCNQ1—Marginal/intermediate cellsKCNJ10—Intermediate cellsSLC2A1—Basal cellsGJA1—ConnexinsGJE1—ConnexinsGJB2—ConnexinsGJB6—Connexins10.1007/s00441-019–03106-7Nordström, CK. et al. (2020)SwedenEndolymphatic sacExperimental studyNANATo analyze Na/K-ATPase and its isoforms in the endolymphatic sac using super-resolution structured illumination microscopyATP1B1—Marginal/intermediate/basal cells10.1002/dneu.22242Pechriggl, EJ. et al. (2015)Austria & SwedenNAExperimental study24NA/Embryos W8-12To analyze the maturation and differentiation of neuronal markers in the fetal cochlea from gestational weeks 8–12PAX2—Marginal cells10.1016/j.ajhg.2022.04.010Trpchevska, N. et al. (2022)VariousHearing LossGenetic association and meta-analysis study723,266Both/60 years oldTo perform a genome-wide association meta-analysis in hearing loss patients to identify the risk loci in cochlear structuresTYR—Marginal/intermediate/basal cellsLMX1A—Marginal/intermediate cellsEYA4—Intermediate cellsABLIM3—Basal cellsGJB2—ConnexinsGJB6—Connexins10.1038/s41598-022–20774-8Zou, J. et al. (2022)China & FinlandMDCase–control study77Both/36–54 years oldTo determine the cytokine profiles of MD and its potential role in autoimmune/autoinflammatory mechanismsHGF—Marginal cells
In the retrieved studies, GJA1, GJB2, and GJB6 were found to be expressed in spiral ligament fibrocytes. Interestingly, GJE1 (known as connexin 23 in mice) was the only connexin identified in the SV, localized in the basolateral membrane of the future marginal cells. However, by embryonic day 16, GJE1 expression was downregulated in marginal cells and was found only in adjacent cochlear cells.
Upon reviewing the reference list of these four records, we found one publication [40] that described three relevant genes to MD and SV; one was expressed in basal cells—GSTM1—and two found in intermediate cells—TMEM176A and TMEM176B. These three genes were found to be related to the immune response in Sporadic MD.
Joint Human and Animal Studies
Both studies involving human and animal subjects (Table 2) were conducted in humans and mice [41–43]. The only gene identified in the two studies was KCNJ10, expressed in SV intermediate cells. Neither of the two studies were related to MD, although they investigated hearing loss and particularly age-related hearing loss (ARHL). Table 2. Summary of selected papers reporting genes in humans and mice (Mus musculus)DOIAuthor/YearCountryDiseaseDesignSample SizeSex/Median AgeMain ObjectiveGene/Protein - ListHuman - Mice (Mus musculus)10.1016/j.heares.2024.109091Chen, J. et al. (2024)UKHearing LossExperimental; Case-controlMice: 3–7; Human: 6099Mice: Both sexes/~14 weeks; Human: NA/44–45 yearsTo investigate how Sgms1 deficiency leads to progressive hearing loss, focusing on endocochlear potential and stria vascularis pathology.Kcnj10 - Intermediate cells10.1113/jphysiol.2006.116889 Knipper, M. et al. (2006)GermanyProgressive High-Frequency Hearing LossMouse Model4-6 cochleae per age groupNA/NATo study Limp2's role in progressive high-frequency hearing loss via Kcnq1/Kcne1 and megalin loss in the stria vascularis.Kcnq1 - Marginal/intermidate cellsKcne1 - Marginal cellsKcnj10 - Intermidate cells10.1016/j.neurobiolaging.2019.04.009Liu, T. et al. (2019)China & USAARHLExperimental; Case-onlyMice:NA; Human: 12Mice: Sexes/1.5-month-old to 2.5 years old; Human: Both sexes/30 to 91 years old.To determine if age-related degeneration of neural crest–derived nonsensory cells in the stria vascularis, outer sulcus cells, and satellite cells in the spiral ganglion, contribute to the development of metabolic and neural forms of age-related hearing lossKcnj10 - Intermediate cells
Animal Studies
Of the 80 mouse model studies analyzed, 52 reported genes expressed in the SV list (Supplementary Table S2). Only seven studies identified Familial MD genes alongside SV marker genes [24, 44–49]. These included two studies on ARHL and one study each on human deafness and head bobbing, hearing impairment, hypothyroidism-related SNHL, hearing loss, and one with no disease associated. The reported Familial MD genes were Myo7a, Hmx2, Otog, Tecta, and Coch. The SV intermediate cell maker Kcnj10 was reported in several studies. In contrast, marginal cell marker genes were described only once—Hspa1b and Atp1b2. Similarly, intermediate cell marker genes—Hsp90b1, Hspa5, and Rorb—and basal cell marker genes—Skp1, Spa1a, Cebpb, S100b, Cdkn1a, Nudt4, and Slc2a1—were reported only once. Furthermore, Pde4b and Kcnq1 were reported in more than one cell marker in the tailored list. Gjb2 was reported once, with expression in cochlear supporting cells and SV basal cells.
Ten rat studies reported genes in the SV (Table 3) [50–59], among them Atp1b2, Slc12a2, Slc2a13, and Kcne1 in the marginal cells; Kcnj10, Slc45a2, and Hif1a in the intermediate cells; and Anxa5, Lamp1, Slc2a1, Mt1a, Arp1a2, and Cldn11 in the basal cells. Cacna1d, Kcnq1, Kcnj13, Atp1b1, and Atp2b1 were reported in two or more SV cell types. In the retrieved studies, Gjb2 was reported in type 1 fibrocytes. None of the studies focused on a particular disease. Similarly, five guinea pig studies found genes Atp1b2 and Slc12a2 in marginal cells, Kcnj10 in intermediate cells, and Rapgef3 and Cldn11 in basal cells, and the described connexins were Gjb2, Gjb3, and Gjb6. Additionally, Pax3 and Kcnq1 were reported as markers for two or more cell types (Table 4) [60–64]. In the retrieved records, Gjb2 was expressed in the basal cells at embryonic day 40 (E40); from E45-E50, it is absent from the cochlea, and from E60 onwards, the expression is found in fibrocytes and spiral ligament. Gjb3 and Gjb6 had no change in expression in the lateral wall. Table 3. Summary of selected papers reporting genes in rats (Rattus norvegicus domestica).DOIAuthor/YearCountryDiseaseDesignSample SizeSex/Median AgeMain ObjectiveGene/Protein - ListRattus norvegicus domestica10.1093/abbs/gms024Chen, J. et al. (2012)ChinaNAExperimental Study6NA/P3-5To investigate the distribution and expression of Cav1.3 channels in the rat cochleaCacna1d - Marginal/Intermediate Cells10.1007/s10571-014-0036-yGross, J. et al. (2014)GermanyNACellular ModelNANA/P3-5To compare the expression of genes involved in different cell death patterns in the stria vascularis, organ of Corti and the modiolus.Hif1a - Intermediate Cells10.1016/j.biocel.2022.106259Huang, S. et al. (2022)ChinaNACellular modelNANA/NATo investigate the role of HIF-1α-mediated autophagy in hypoxic cochlear marginal cells and its protective mechanism against apoptosisHif1a - Intermediate CellsAnxa5 - Basal Cells10.1080/03655230701624830Gu Hur, D. et al. (2007)KoreaNAExperimental Study12Female/W7To determine whether skeletal muscle denervation causes post-transcriptional mechanisms to regulate AChR β-subunit mRNAs.Kcnq1 - Marginal/Intermediate CellsKcne1 - Marginal Cells10.1038/srep20903Liu, J. et al. (2016)ChinaNACellular modelNANA/NATo identify the cell organelles that store ATP in cochlear marginal cellsLamp1 - Basal Cells10.1016/j.heares.2011.03.011Mazurek, B. et al. (2011)GermanyNACellular model18NA/P3-P5To investigate the expression of genes involved in oxidative stress response in cochlear tissues and to explore their role in cochlear injury during the developing period under different oxygen conditions.Slc2a1 - Basal CellsMt1a - Basal Cells10.1038/s41598-018-38079-0Nonomura, Y. et al. (2019)JapanNAExperimental Study64Male/W7To profile and structurally characterise N-glycans in the stria vascularis to see how they are involved in regulating membrane activity and secreted proteins.Cldn11 - Basal Cells10.1152/physiolgenomics.00006.2005Pondugula, S.R. et al. (2006)USANACellular modelNANA/NATo identify the genes involved in Na⁺ transport in rat semicircular canal duct epithelium and determine how their expression is regulated by glucocorticoids.Kcnj13 - Marginal/Intermediate CellsKcnj10 - Intermediate Cells10.1016/j.neures.2013.06.003Takiguchi, Y. et al. (2013)JapanNAHistological; immunohistochemical12NA/W6-W8To investigate the long-term effects of mitochondrial dysfunction induced by 3-nitropropionic acid on cochlear fibrocytes and ion transport proteins in the cochlea, and examine changes in the expression of key proteins in response to cochlear degenerationSlc12a2 - Marginal CellsKcnj10 - Intermediate CellsGjb2 - Connexins10.1111/ejn.12973Uetsuka, S. et al. (2015)JapanNAExperimental109NA/W7To identify proteins involved in ion and organic substance transport in the stria vascularis and discover new candidates for deafness-related genes.Atp1b1 - Marginal/Intermediate/Basal CellsKcnj13 - Marginal/Intermediate CellsAtp2b1 - Marginal/Intermediate CellsKcnq1 - Marginal/Intermediate CellsKcne1 - Marginal CellsSlc2a13 - Marginal CellsSlc12a2 - Marginal CellsAtp1b2 - Marginal CellsSlc45a2 - Intermediate CellsKcnj10 - Intermediate CellsSlc2a1 - Basal CellsAtp1a2 - Basal CellsTable 4Summary of selected papers reporting genes in guinea pigs (Cavia spp.), birds (Gallus gallus domesticus and Tyto alba guttata) and frogs (Xenopus laevis)DOIAuthor/YearCountryDiseaseDesignSample SizeSex/Median AgeMain ObjectiveGene/Protein—ListCavia spp.10.3389/fnmol.2022.842132Edvardsson Rasmussen, J. et al. (2022)SwedenNAExperimental18Both/W6-9To investigate the time-dependent effects of acute furosemide administration by localizing Fetuin-A and PEDF proteins in the guinea pig cochleaSlc12a2—Marginal cells10.1007/s00441-006–0369-zJin, Z, et al. (2007)SwedenHereditary deafnessExperimentalNANA/varied from E35 to adultTo investigate the molecular basis of hereditary deafness in the German waltzing guinea pigPax3—Marginal/intermediate cellsAtp1b2—Marginal cellsKcnj10—Intermediate cellsGjb2—ConnexinsGjb3—ConnexinsGjb6—Connexins10.1111/j.1460–9568.2007.05994.xJin, Z, et al. (2008)SwedenHereditary deafnessExperimentalNANA/E35-P0To examine the developmental patterns of key potassium transport proteins in the cochlear lateral wall of German waltzing guinea pigs with hereditary deafnessSlc12a2—Marginal cellsCldn11—Basal cells10.1007/s00405-022–07380-0Wang, C. et al. (2022)ChinaNAExperimental30NA/NATo investigate the expression and localization of Epac1 and Epac2 in the guinea pig inner ear and their potential role in inner ear microcirculationRapgef3—Basal cells10.1007/s00405-014–3093-4Xiong, M. et al. (2014)ChinaNAExperimental30NA/NATo determine whether Radix astragali can prevent or reduce the down-regulation of connexin 26 in the Stria vascularis of the guinea pig cochlea following acoustic traumaKcnq1—Marginal/intermediate cellsGallus gallus domesticus & Tyto alba guttata10.1038/srep34203Wilms, V. et al. (2016)GermanyNAExperimentalNANA/varied from P18 to P966To investigate the molecular bases of K + secretory cells in the inner ear in birds and mammalsKcnj10—Intermediate cellsAtp1a2—Basal cellsGjb2—ConnexinsGjb6—ConnexinsXenopus laevis10.1159/000356353Parrock, S. et al. (2013)UKEAST SyndromeCase-onlyNANA/NATo characterize Kcnj10 mutations in patients with EAST syndrome to investigate the role of Kcnj16 co-expression in modulating Kcnj10 channel functionKcnj10—Intermediate Cells
On the other hand, one study performed on birds had genes KCNJ10 and ATP1A2 from intermediate and basal cells, respectively, and the described connexins—GJB2 and GJB6—were not found expressed in the SV [65]. The paper that described several connexins in the guinea pig was focused on hereditary deafness, whereas the study that utilized birds as a model was performed in wild-type animals. The X. laevis study only identified the intermediate cell gene KCNJ10 while investigating Epilepsy, Ataxia, Sensorineural deafness, and Tubulopathy (EAST) syndrome [66].
Risk of Bias Assessment
Human and animal risk of bias analyses are summarized in Supplementary Tables S4 – S6.
Audiovestibular and Immune Phenotype in Mouse Knockout of Stria Vascularis Genes
Knocked out Gja1 generated mice had significant immune dysregulation with an abnormal spleen morphology. Furthermore, Kcne1-knockout mice had an increased number of monocytes and neutrophils and a decreased number of lymphocytes, as well as severe hearing loss across all frequencies (Table 5). Table 5. Audiovestibular and immune response phenotype information from the International Mice Phenotyping Consortium for the selected SV genesGeneAudiovestibularImmune responseActn1NSNSQsox1NSNSMdh1NSNSActb––Kcne1SignificantSignificantEsrrb––Cacna1d––Coll11a2NSNSSlc12a2––Hgf––Gstm1––Gjb2––Gjb6–NSGja1NSSignificantGje1NSNSTmem176aNS–Tmem176b––NS not significant
Familiar MD Gene Expression in the Stria Vascularis
Three single-cell preservation methods from the lateral wall dataset in the gEAR portal: fresh tissue, RNA-later, and methanol fix, were used. The only gene that was found consistently across all three preservation methods was Coch, which had high expression in fibrocytes with 2.08 logFC and present in 63% of cells (fresh tissue), 1.78 logFC and found in 70% of cells (RNA-later), 1.41 logFC and 65% of cells (methanol fix) (Table 6). On the other hand, Dtna was found highly expressed in fresh tissue Reissner’s cells (1.10 logFC in 49% of cells) and in RNA-later spindle cells (1.05 logFC in 70% of cells). Prkcb was found in immune cells: B cells in fresh tissue (1.23 logFC in 51% of cells) and macrophages in RNA-later and methanol fix (1.00 logFC in 60% of cells and 1.06 logFC in 68% of cells, respectively). Furthermore, Gjd3 was not found in any of the cells in the dataset. Table 6. Average expression of Familial MD genes in the lateral wall from Gu et al. (2020) found in the gEAR portalMethodSpindle/rootFibrocytesReissner’sB cellsMacrophagesFresh tissue–Coch (N = 30, 63.33%, mean: 2.08)Dtna (N = 51, 49.02%, mean: 1.10)Prkcb (N = 142, 51.41%, mean: 1.23)–RNA-laterDtna (N = 54, 70.37%, mean: 1.05)Coch (N = 74, 70.21%, mean: 1.78)––Prkcb (N = 15, 60.00%, mean: 1.00)Methanol fix–Coch (N = 101, 65.35%, mean: 1.41)––Prkcb (N = 25, 68.00%, mean: 1.06)
Gene Set Enrichment Analysis
Gene Set Enrichment Analysis was performed using “human” as species on candidate genes: ACTN1, QSOX1, MDH1, ACTB, KCNE1, ESRRB, CACNA1D, COL11A2, SLC12A2, and HGF (Fig. 2),2 and connexins GJA1, GJB2, GJB6, and GJE1. The resulting active pathways were not specific to the inner ear auditory and vestibular dysfunction, nor immune dysregulation. The top three enriched pathways for GO BP (Supplementary Table S7) were “cell communication by electrical coupling” (adjusted p-value [adjusted p-val] = 1.61 × 10^−6^; Relative Enrichment [RE] = 558), “maintenance of blood–brain barrier” (adjusted* p*-val = 1.61 × 10^−6^; RE = 153), and “gap junction assembly” (adjusted* p*-val = 1.71 × 10^−6^; RE = 488). For GO CC (Supplementary Table S8) and GO MF (Supplementary Table S9), the top two enriched pathways related to gap junction activity were “connexin complex” (adjusted* p*-val = 1.11 × 10^−7^ RE = 260), and “gap junction” (adjusted* p*-val = 7.17 × 10^−5^ RE = 132), and “gap junction channel activity” (adjusted* p*-val = 7.57 × 10^−8^; RE = 238), and “gap junction channel activity involved in cell communication by electrical coupling” (adjusted* p*-val = 7.57 × 10^−8^; RE = 982).Fig. 2. Representation of the stria vascularis (SV) and MD genes found in the lateral wall of the scala media
For KEGG (Supplementary Table S10), the three principal pathways were “arrhythmogenic right ventricular cardiomyopathy” (adjusted* p*-val = 1.05 × 10^−2^; RE = 29), “Focal adhesion” (adjusted* p*-val = 3.35 × 10^−2^; RE = 12) and “Tight junction” (adjusted* p*-val = 3.35 × 10^−2^; RE = 15).
No transcription factors (TF) were significant (Supplementary Table S11). It is worth noting that the gene count was always below 50% in the GO, KEGG, or collecTRI analyses.
Discussion
Menière’s Disease is a multifactorial disease with genetic contribution in 20–30% of cases [15]. In this review, we analyzed studies published over the past 20 years that investigated gene expressions in SV marginal, intermediate, and basal cells, and the relevance to MD. These studies included both human and animal models and described over 20 genes in total. Among these, 13 SV-associated genes—ACTB, ACTN1, CACNA1D, COL11A2, ESRRB, GSTM1, HGF, KCNE1, MDH1, QSOX1, SLC12A2, TMEM176A, and TMEM176B—and gap junction proteins were consistently identified as being particularly important in the context of MD pathogenesis.
Marginal Cells
Marginal cells are the innermost cell type of the SV and are in direct contact with the scala media [67]. Their primary function is the active transport of potassium ions (K^+^) from the other outer SV cell types to cochlear duct to maintain endocochlear potential [68]. We found that marginal cell markers QSOX1 and MDH1 were reported in two independent MD studies [38, 40]. The sulfhydryl oxidase 1 protein (QSOX1 gene) is a crucial disulfide bond-forming enzyme that facilitates integration of laminin into the extracellular matrix, for the matrix ability to support integrin-dependent cell adhesion and migration [69]. Although the role of QSOX1 encoded protein in audiovestibular dysfunction is yet to be elucidated, studies have found that QSOX1 may be involved in the immune response exerting anti-inflammatory effects in sepsis, by inhibiting M1 macrophage polarization. Furthermore, it regulates inflammatory signaling by preventing epidermal growth factor receptor activation and promoting ubiquitin-mediated regulation [70]. On the other hand, malate dehydrogenase, cytoplasmic protein (MDH1 gene) is a NAD(H)-dependent enzyme [71], and evidence of its function on the inner ear is scarce. The MDH1-encoded protein is part of the malate-aspartate shuttle (MAS), crucial to maintaining intracellular NAD(H) redox balance by transferring reduced equivalents across the impermeable inner mitochondrial membrane, as NAD(H) cannot cross it [72]. A homozygous variant in MDH1 (p.Ala138Val) has been linked to developmental delay, epilepsy, and progressive microcephaly [73]. Conversely, MDH1 can drive functional impairment to CD^8+^ T cells in patients with triple-negative breast cancer, promoting their differentiation to a CD^8+^ T exhausted phenotype, which may disrupt immune balance within the breast cancer environment [74].
The potassium voltage-gated channel subfamily E member 1 protein, encoded by KCNE1 gene, functions by dimerizing with KCNQ1-encoded protein to modulate the channels K^+^ secretion to the cochlear and vestibular endolymph [75–77]. Homozygous or compound heterozygous variants in the KCNE1 and KCNQ1 have been associated with the Jervell and Lange-Nielsen syndrome type 2 [78–80], characterized by cardiac arrhythmias and increased risk of sudden death, and associated with severe bilateral sensorineural hearing loss. In the tailored list, both ESRRB and CACNA1D were found to be upregulated in marginal cells but downregulated in intermediate cells. ESRRB encodes the steroid hormone receptor ERR2 protein, and CACNA1D encodes the voltage-dependent L-type calcium channel subunit alpha-1D. Both proteins have been implicated in hearing loss–related disorders, such as DFNB35 and familial sinus node dysfunction [81–85]. However, none of the cases with these disorders have presented vestibular dysfunction, commonly observed in MD patients.
Solute carrier family 12 member 2 protein is encoded by SLC12A2 gene. Its main function is to transport Cl^−^, K^+^, and/or Na^2+^ across the plasma membrane [86]. Research on the stria vascularis has reported that variants in the carboxy-terminal domain of SLC12A2-encoded protein are responsible for hereditary hearing loss across all frequencies. The study also reported that some of the patients with these mutations had vestibular impairments [87]. Furthermore, SLC12A2 has been implicated in the immune system, being critical for apoptotic cell uptake. The study found that deficiency of SLC12A2 increased efferocytosis in phagocytes and macrophages, exacerbating the inflammatory response [88].
HGF gene encodes for hepatocyte growth factor, and it has been linked directly to the immune response in MD cases [6, 37]. Hepatocyte growth factor receptor is encoded by the MET gene, and it is expressed in the cell’s surface [89]. HGF/MET signaling has been involved in immunoregulating processes such as monocyte-macrophage polarization, dendritic cell migration and deactivation, and B cell adhesion [89–92]. In the context of MD, the HGF-encoded protein—along with granulocyte colony-stimulating factor (G-CSF) and IL-8—has been hypothesized to contribute to the formation of neutrophil extracellular traps, contributing to the inflammatory response [37]. Furthermore, a single-cell RNA sequencing study found that patients with an autoinflammatory MD phenotype exhibit an active population of monocytes characterized by high transcript levels of IL-1β receptors and IL-8. The study found that the monocytes could be polarized by a combination of HGF, vascular endothelial growth factor (VEGF), and stem cell factor (SCF) [6]. Besides its involvement in the immune system, it has been reported that noncoding mutations in hepatocyte growth factor have been associated with DFNB39 [93].
Intermediate Cells
ACTB encodes protein actin, cytoplasmic 1, also known as beta-actin. Beta-actin is an important structural protein, and it is expressed in almost all adult human cell types [94, 95]. Mutations in ACTB-encoded protein have been linked to an array of diseases ranging from deafness to lymphomas [96, 97]. Several studies have found that mutations in the ACTB gene lead to the Dystonia-Deafness syndrome, characterized by sensorineural hearing loss and dystonia [96, 98].
Intermediate cells are neural crest-derived melanocytes that form the middle layer of the SV [99]. They contribute to the supporting function of the cochlear blood labyrinth barrier (BLB), which strictly regulates the flow of ions and metabolites from the capillary network to the endolymph [99]. There is a link to congenital deafness in intermediate cells, as the deficiency of these cells results in a low endocochlear potential that is insufficient to reach the threshold of compound action potential through sound pressure [100]. In a previous study, researchers were able to identify marker genes TMEM176A and TMEM176B, which are associated with the intermediate cells of the SV and are also associated with MD [40, 101]. Most of the studies examined the co-regulation and interaction of TMEM176A and TMEM176B, given their structural similarities and shared involvement in intracellular dynamics. Transmembrane protein 176 A (TMEM176A gene) and Transmembrane protein 176B (TMEM176B gene) are homologous genes that suppress the maturation of dendritic cells [102]. Although in vivo studies of these genes remain largely unknown, previous knockout mouse models found that TMEM176A/B is functionally involved in the MHC class II presentation by regulating the type-2 conventional dendritic cells (cDC2) [103]. Furthermore, the proteins encoded by TMEM176A/B localize in the Golgi apparatus in the late endolysosomal system and colocalize with HLA-DM, which catalyzes peptide exchange on MHC class II molecules [103, 104]. Although there are limited studies that cater TMEM176A functionality, TMEM176B in particular has also been known to mediate Na + efflux for facilitating charge compensation for V-ATPase driven acidification [102, 105]. This process regulates phagosomal pH, which is critical for antigen cross-presentation to dendritic cells [105]. Additionally, other knockout studies of TMEM176B have found defective functionality in cerebellar granulocytes, which resulted in the development of sporadic ataxia [102].
Basal Cells
Basal cells are the outermost layer of the SV that forms a critical barrier between the endolymph and intrastrial space [100]. They connect to the fibrocytes of the spiral ligament, allowing them to regulate ion exchange into the SV while preventing leakage between cochlear compartments [99]. In the study, we identified GSTM1 and ACTN1 as basal cell markers for the SV. Glutathione S-transferase Mu 1 (GSTM1 gene) is a detoxification enzyme that catalyzes the conjugation of reduced glutathione (GSH) to electrophilic compounds [106]. They also serve a protective role against carcinogens such as polycyclic aromatic hydrocarbons (PAH), suggesting that the deficiency of this gene increases the risk of cancer [107]. Aside from xenobiotic metabolism, GSTM1 is involved in modulating cell signaling by conjugating prostaglandins A_2_ and J_2_ (PGA_2_ and PGJ_2_, respectively). This enzymic activity mitigates the antiproliferative effects of these prostaglandins, suggesting that it may play a role in cell proliferation and inflammatory process [108]. Additionally, GSTM1 modifies lipid signaling molecules by catalyzing 14,15-Hepoxilin A_3_ (14,15-HxA_3_) to a cysteinyl derivative (14,15-HxA_3_) [109]. On the other hand, Alpha-actinin-1 (ACTN1 gene) is a cross-linking protein for filamentous actin (F-actin) that contributes to cytoskeletal organization and cell adhesion [110]. Abundantly expressed in the smooth muscle cells, ACTN1 is essential for T-cell migration and functionally interacts with ICAM-1 to facilitate leukocyte migration to the surrounding tissues [111]. It also binds to CLP36, forming a complex that associates with actin filaments and stress fibers in activated platelets and endothelial cells [112]. ACTN1 is found in the stereocilia of hair cells where it plays a critical role in actin-binding activity that is regulated by Ca^2+^ through MET channels [113, 114]. Although it is expressed at low levels in the auditory and vestibular stereocilia, this process forms an essential part of stabilizing the stereocilia cytoskeleton [113, 114].
Collagen alpha-2(XI) chain is encoded by the COL11A2 gene. The role of COL11A2 in the inner ear is not well known; however, several collagen proteins are expressed within the cochlea [115]. A study on Zebrafish identified that COL11A2 is essential in maintaining the properties of the cartilage matrix [116]. Several studies have reported that mutations in the COL11A2 gene lead to non-syndromic and syndromic deafness [117, 118]. Research has shown that mutations in COL11A2 can lead to syndromes that may have concurrent vestibular impairments, such as Stickler Syndrome [119].
Connexins
Gap junction proteins are a protein family essential in epithelial intracellular junctions [120], as they form hemichannels in the plasma membrane from hexameric structures, known as a connexon [121]. The union of two connexins between two cells is known as a gap junction, which allows for intracellular exchange of molecules, such as Ca^2+^, ATP, glutamate, or NAD^+^ [122]. In this review, we found two connexins expressed in the SV—GJB2 and GJE1. The gap junction beta-2 protein (GJB2 gene) has been identified in non-syndromic SNHL, with three main mutations c.35delG driving 40–70% of deafness cases in the European, North African, Middle Eastern, Sub-Saharan African, North and South American, and Australian populations [123, 124]. On the other hand, c.235delC and c.109G > A variants are more frequently found across several East Asian populations [121]. Gap junction epsilon-1 protein (GJE1 gene) role in hearing loss is less studied. A study found 13 (5.14%) of 253 unrelated Taiwanese patients with nonsyndromic hearing loss had variants in GJE1 [125]. Although GJB2 and GJE1 were the only two connexins after filtering, GJA1, GJB3, and GJB6 were present in some of the selected studies.
Gap junction alpha-1 protein (Gja1 gene) knockout mice, generated by the IMPC, were found with abnormal spleen morphology. Although no audiovestibular phenotype was recorded by the IMPC, the GJA1-encoded protein is essential in humans, and mutations in the gene have been found to contribute to nonsyndromic hearing loss [126]. Furthermore, a study found that suppressing Gja1 in mice creates hyperpermeability in the SV, decreasing endocochlear potential, leading to mild hearing loss [127].
The importance of connexins in MD has yet to be fully elucidated; however, several variants forming a haplotype in gap junction delta-3 protein (GJD3 gene) have been found in both sporadic and Familial MD, suggesting that rare variation in connexins can lead to audiovestibular dysfunction [16].
Stria Vascularis and Menière’s Disease
Besides the SV marker genes used in this study to screen previously published papers, an independent study found 181 genes enriched in Sporadic MD and 264 genes in Familial MD, with 43 genes shared between both MD subsets [12], among them the KIF1B gene. A variant in the KIF1B gene has been found to possibly contribute to pathogenesis in the autoinflammatory phenotype in some Sporadic MD patients [128]. None of the Familial MD genes showed to be enriched in the single-cell RNA sequencing SV dataset, although we did find enrichment of Prkcb in B cells and macrophages. A mutation in a conservative serine phosphorylation site in the protein kinase C beta type (PRKCB gene) has been associated with increased tyrosine phosphorylation and membrane association of Bruton's tyrosine kinase and increased signaling through the B-cell receptor (BCR) in B cells and the high-affinity IgE receptor in mast cells. Furthermore, PRKCB inhibition causes an overexpression of Ca^2+^ signaling triggered by BCR [129].
A study published in 2025 using single-cell RNA sequencing on SV whole-tissue explants from neonatal and mature mice found that TNF-like Weak inducer of apoptosis (TWEAK) was released from the intermediate cells of the SV to the marginal cells and spindle cells, where its receptor tumor necrosis factor receptor superfamily member 12 A (TNFRSF12A gene) was expressed [18]. Additionally, a study on MD reported that patients with the allelic variant rs4947296 (chr6:31014645T > C) showed 973 differentially expressed genes, including NF-kappa-B p105 subunit (NFKB1 gene) and TNFRSF12A [130]. Furthermore, carriers of the CC risk genotype had higher expression of both TNFRSF12A and NFKB1 compared to the TT protective genotype. When haplotype-conditioned lymphocytes were stimulated with TWEAK, the CC genotype had no significant increase in expression of TNFRSF12A; conversely, NFKB1 was significantly upregulated when compared to the stimulated TT genotype. This allelic variant is an expression quantitative trait locus that regulates the TWEAK pathway, leading to the activation of NF-κB inflammatory pathway in MD patients [130].
Limitations
This systematic review has its limitations. First, the SV marker gene list was generated from P30 mice and described MD genes in humans. Second, the number of SV studies in MD patients or animal models is low. Third, most of the studies lacked information on genetic variants and their effects on the inner ear. Additionally, some of the studies exhibited moderate to high risk of bias; thus, results should be interpreted with caution. Finally, while animal models were included, protein function often differs across species, limiting the extension of findings to MD patients.
Conclusions
Some genes found in the SV may contribute to the auditory dysfunction in MD, but most genetic variants may be involved in the immune response commonly found in Sporadic MD cases. The role of gap junction proteins in the SV and their connection to MD pathophysiology is yet to be elucidated.
Supplementary Information
Below is the link to the electronic supplementary material.ESM 1(XLSX 118 KB)