Biodiversity of strains belonging to the freshwater genus Aquirufa in a riparian forest restoration area in Salzburg, Austria, with a focus on the description of Aquirufa salirivi sp. nov. and Aquirufa novilacunae sp. nov
Alexandra Pitt, Stefan Lienbacher, Johanna Schmidt, Meina Neumann-Schaal, Jacqueline Wolf, Hannah Wenng, Aharon Oren, Zoe Huber, Martin W. Hahn

TL;DR
This study discovered two new species of freshwater bacteria, Aquirufa salirivi and Aquirufa novilacunae, in a riparian forest in Austria and analyzed their genetic and ecological characteristics.
Contribution
The paper describes two new Aquirufa species and provides insights into their genomic and ecological traits, including their distribution in global water samples.
Findings
Aquirufa salirivi sp. nov. was not detected in public metagenomes, suggesting it may be a rare or newly emerging species.
Aquirufa novilacunae sp. nov. was found in 15 water samples globally, mainly in rivers from Asia, North America, and Australia.
Genomic analyses confirmed the two new species based on average nucleotide identity thresholds and distinct gene content.
Abstract
During a citizen science project, four freshwater habitats in a riparian forest restoration area in Salzburg, Austria, were sampled. The primary aim was to obtain bacterial strains of the genus Aquirufa, a group of typical and widespread freshwater bacteria. Numerous pure cultures of Aquirufa strains could be obtained, three of them originating from the river Salzach, a newly created pond and the lake Ausee represented new species. Strain 1-SAACH-A3T was characterized by a genome size of 3.2 Mbp and a G + C value of 38.4 mol% and encoded genes predicted for nitrate uptake and nitrous oxide utilization. Strains BAHN-186BT and 2-AUSEE-184A6 were characterized by a genome size of 2.4 Mbp and a G + C value of 42.4 and 42.2 mol%, respectively, and encoded genes predicted for the light-harvesting rhodopsin system. Calculated whole-genome average nucleotide identity values with Aquirufa type…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1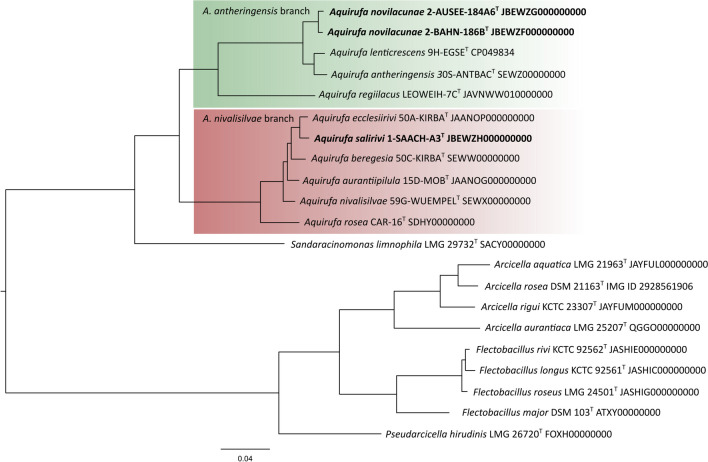 Figure 2
Figure 2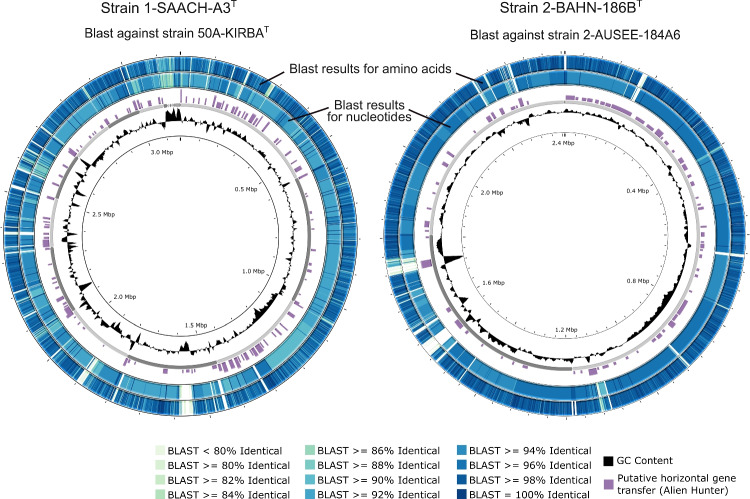 Figure 3
Figure 3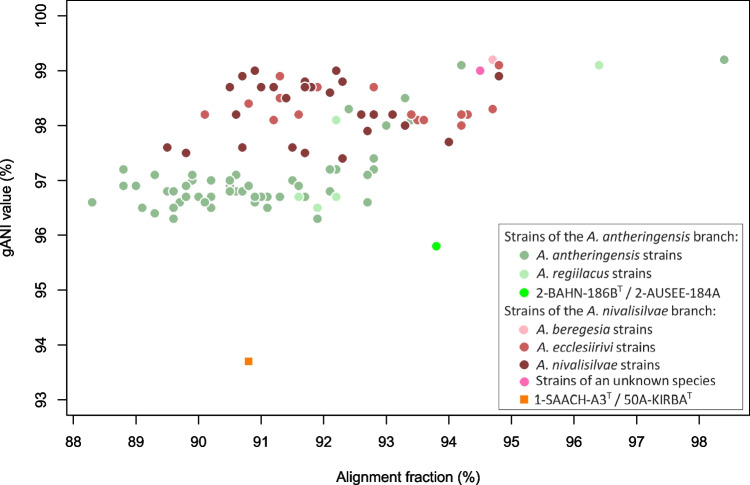 Figure 4
Figure 4- —https://doi.org/10.13039/501100013699Bundesministerium für Bildung, Wissenschaft und Forschung
- —https://doi.org/10.13039/501100004955Österreichische Forschungsförderungsgesellschaft
- —University of Innsbruck and Medical University of Innsbruck
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Microbial Community Ecology and Physiology · Protist diversity and phylogeny
Introduction
The genus Aquirufa was established in 2019 (Pitt et al. 2019) and encompasses currently eight species with validly published names (Pitt et al. 2019, 2020, 2022, 2024; Sheu et al. 2020). The genus represents a group of typical and widespread freshwater bacteria. It belongs to the order Cytophagales (phylum Bacteriodota) and was originally assigned to the family Cytophagaceae (Pitt et al. 2019). Whole-genome-based phylogenetic analyses showed polyphyly of the family Cytophagaceae (García-López et al. 2019), and some authors proposed a reorganization of the taxon. Currently, the situation remains unsatisfactory, because the LPSN Database (Parte 2013) assigns the genus Aquirufa to the family Spirosomataceae while the NCBI Taxonomy database (Schoch et al. 2020) follows Lu et al. (2024) who created the new family Flectobacillaceae including the genus Aquirufa along with four further genera. Genome-based phylogenetic trees showed that the genus Aquirufa forms two distinct branches, the A. antheringensis branch and the A. nivalisilvae branch (Pitt et al. 2024). The species of the A. antheringensis branch have slightly smaller genome sizes and higher genomic G + C values than the species of the A. nivalisilvae branch. Typical for all Aquirufa strains are genome sizes around 3 Mbp and genomic G + C values around 40 mol%, red pigmentation, and rod-formed cells; they grow aerobically and chemoorganotrophically.
Within a citizen science project, freshwater habitats in a riparian forest restoration area in Salzburg, Austria, were sampled over 1 year with the primary aim of obtaining bacterial isolates belonging to the genus Aquirufa. This area called “Weitwörther Au” is a part of the riparian area of the river Salzach (Salzachauen) which is included in the Natura 2000 network of protected areas. It is located north of the city of Salzburg and covers an area of 3 km^2^. From 2015 to 2021, the Weitwörther Au was the focus of a renaturation project, aiming to reestablish natural conditions (Land-Salzburg 2021). Historically, this riparian area was used for intensive agriculture, forestry (specifically spruce monoculture), and gravel and sand extraction. Approximately 20 years ago, sand mining led to the creation of an artificial lake, the Ausee, which had been used for fishing. The main goal of the renaturation was to recreate various typical natural habitats through the conversion of forests, the establishment of ponds fed by groundwater and precipitation and the revitalizing of the water bodies like tributaries of the Salzach and the quarry lake.
The sampling of four selected water bodies led to several pure cultures assigned to the genus Aquirufa. Besides strains belonging to known Aquirufa species, some isolates represented new species. Strain 1-SAACH-A3^T^ originated from a water sample taken from the river Salzach in the summer of 2023 and 2-BAHN-186B^T^ and 2-AUSEE-184A6 from water samples taken in autumn 2023 in the restoration area beside the river from a small pond and the lake, respectively. Genome sequencing and calculated whole-genome average nucleotide identity values (gANI) revealed that strain 1-SAACH-A3^T^ represents a new species within the A. nivalisilvae branch, for which we propose the name Aquirufa salirivi sp. nov. Strains 2-BAHN-186B^T^ and 2-AUSEE-184A6 belonging to the A. antheringensis branch of the genus were of special interest. Calculated overall genome-related indexes were very close to demarcation values separating two species but revealed that the two strains represent only a single new species, for which we propose the name Aquirufa novilacunae sp. nov. with 2-BAHN-186B^T^ as the type strain.
Materials and methods
Sampling and isolation of strains
Within the riparian forest restoration and wetland area “Weitwörther Au,” we chose four sampling sites, the river Salzach, the creek Reitbach, the lake Ausee, and a newly created small pond. Figure 1 shows the study area and was generated using SAGIS a geographical information system provided by the province/federal state of Salzburg (https://www.salzburg.gv.at/sagis). Water samples were taken from approximately 0.1-m depth, and measurements for pH, electrical conductivity, water temperature, oxygen concentration, and saturation were performed on location with a WTW multiparameter probe. The water samples were filtered as soon as possible through 0.65-μm pore size filters to exclude larger bacterial cells and the obtained filtrates were subsequently spread on nutrient broth-soytone-yeast extract (NSY) (Hahn et al. 2004) agar plates and incubated at room temperature (about 21 °C). The agar plates were observed for several weeks. Growing colonies with the typical red pigmentation of strains belonging to the genus Aquirufa were picked and transferred to 24 well plates filled with liquid NSY medium. Promising cultures showing the expected pigmentation were transferred to Erlenmeyer flasks and purified by alternating culturing in a liquid medium and on agar plates. Because the 16S rRNA gene is in the case of the genus Aquirufa not suitable to distinguish between species (Pitt et al. 2024), we used as a marker the partial sequences of the gyrB gene encoding for the B subunit of the DNA gyrase (Pitt et al. 2022) and a threshold of 97% to assign the strains to a known species (Pitt et al., in revision). In addition, the strains which likely represent a new species were genome-sequenced and gANI values were calculated. All strains were stored at − 80 °C in a liquid NSY medium supplemented with 15% glycerol.Fig. 1. Map of the sampled habitats in the riparian forest restoration area Weitwörther Au. 1, river Salzach; 2, newly created pond; 3, lake Ausee; 4, creek Reitbach
Whole-genome sequencing and analyses
DNA extraction and genome sequencing were performed following the method described previously (Hoetzinger et al. 2017). A shotgun library was paired-end sequenced on an Illumina NovaSeq sequencer with 2 × 150 bp. For the de novo assembly of the genomes served SPAdes version 3.13.1 (Bankevich et al. 2012). The three genomes described here were deposited in DDBJ/ENA/GenBank databases and annotated by the NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al. 2016). In addition, the genomes were annotated by the Integrated Microbial Genomes & Microbiomes Expert Review (IMG/MER) system (Chen et al. 2019) and added to the IMG/MER database. The IMG/MER system served also for calculating gANI values and corresponding alignment fraction (AF) values for all possible pairwise comparisons of the new strains with type strains of previously established Aquirufa species. Digital DNA-DNA hybridization (dDDH) values were calculated in addition using the Type (Strain) Genome Server (Meier-Kolthoff et al. 2021) for the new strains and closely related type strains. For visualization of the genome sequences, we used the tools Proksee (Grant et al. 2023), BLAST (Altschul et al. 1990) and Alien Hunter (Vernikos and Parkhill 2006). Besides the genome sequences of the three strains described in this study, we used 36 further previously established genome sequences of Aquirufa strains (Table S1), which were almost exclusively isolated from freshwater habitats in Austria, for calculating intra-species gANI and AF values with the IMG/MER system. For a graphical representation of the values served R (RCoreTeam 2020) and RStudio (RStudioTeam 2019).
Phylogenetic reconstructions
For phylogenetic reconstructions, we considered besides all type strains of the genus Aquirufa, type strains of the closely related genera Sandaracinomonas, Pseudarcicella, Arcicella, and Flectobacillus with available genome sequences.
A phylogenetic tree based on almost full-length 16S rRNA gene sequences was calculated with the software Mega X (Kumar et al. 2018). All sequences were aligned, trimmed, and utilized for the construction of a neighbor-joining tree with 1000 replicates based on the Kimura 2 parameter model (Kimura 1980) with the settings gamma-distributed (1 gamma Parameter) and gaps pairwise deleted. A genome-based phylogenetic tree was calculated by the use of amino acid sequences of the core genes. Therefore, all used genome sequences were annotated by Prokka version 1.14.6 (Seemann 2014) with the standard settings. The core genes of the sequence set were identified with the tool Roary version 3.13.0 (Page et al. 2015), with the setting of 65% minimum protein sequence similarity and the requirement that a gene/protein has to occur in all genomes. For both tools, we utilized The European Galaxy server (Galaxy-Community 2024). The obtained sequence set of 658 core genes was translated into protein sequences with Mega X (Kumar et al. 2018) and aligned by MAFFT (Katoh et al. 2005). The tool GBLOCKS, version 0.91b (Castresana 2000) served to filter out highly variable positions, which reduced the primary alignment from 247,753 to 228,366 alignment positions (92%) in 1164 selected blocks. A RAxML tree (Stamatakis 2014) was constructed on the platform CIPRES Science Gateway version 3.3 (Miller et al. 2010), with standard settings and 100 bootstrap replicates.
Metagenome sequencing and analyses
Metagenome sequencing was performed with the water sample of the Ausee from November 2023. Approximately 300 ml of sampled water was prefiltered through 0.8-μm pore size filters to exclude larger particles and large bacterial cells and to enrich smaller ones like the target bacteria. The filtrate was subsequently filtered onto 0.2-μm pore size filters. DNA was extracted using phenol–chloroform-isoamyl alcohol as reported by Schauer et al. (2003). The library was sequenced on an Illumina NovaSeq with sequence mode NovaSeq PE 150, which resulted in 8.4 Gbp in 28.2 million reads. The metagenome as well as 123 publicly available metagenomes from freshwater habitats obtained from locations all over the world were analyzed by mapping as follows: We prepared a set of concatenated genome sequences with removed ribosomal operons of all Aquirufa type strains including 1-SAACH-A3^T^ and 2-BAHN-186B^T^. On the European Galaxy server (Galaxy-Community 2024), the reads of the metagenomes were mapped on the genome set with Bowtie2 (Langmead and Salzberg 2012) with settings selectively mapping only reads with whole-length (option end-to-end) and sequence identities of ≥ 95% reflecting the species demarcation threshold. Subsequently, the coverage for every base of the genome set was obtained by the use of the software Bedtools (Quinlan and Hall 2010). The data were analyzed with R (RCoreTeam 2020) and RStudio (RStudioTeam 2019). Infrequent occurring species could cause results characterized by low coverage breadth combined with low coverage depth, potentially indicating that the number of sequenced base pairs limited the detection of the reference genome. So, we used the following analysis to create a test criterion for distinguishing between detection and no detection. Metagenomes harboring genomes of Polynucleobacter paneuropaeus (i.e., mapping results with coverage depth > 100-fold and breadth > 70%) were separately mapped on genome sequences of five Polynucleobacter paneuropaeus strains (ANI > 98% but differing in their sets of accessory genes). The number of mapped metagenomic reads was stepwise reduced and the respective coverage depth and breadth were recorded. The plotted results showed for all five Polynucleobacter paneuropaeus genomes asymptotic curves (Fig. S1). The genome-specific data set with the smallest asymptote (reflecting the size of the core genome) was used to model the curve (Fig. S1). The obtained function (Fig. S1) was used to test whether a certain mapping result must be considered as detection. If the coverage breadth obtained by mapping metagenomic reads is at least tenfold and larger than the calculated coverage breadth (Fig. S1), a particular species is supposed to be detected. The obtained metagenomic reads from the Ausee sample were assembled by the use of metaSPAdes 3.15.3 (Nurk et al. 2017) and merged into metagenome-assembled genomes (MAGs) with MetaBat2 (Kang et al. 2019). CheckM lineage_wf (Parks et al. 2015) and GTDB-Tk Classify genomes (Chaumeil et al. 2019) were used for quality check and taxonomic classification, respectively. All tools were utilized on the European Galaxy server (Galaxy-Community 2024). GANI values were calculated with the ANI Calculator on Ezbiocloud (Yoon et al. 2017).
Phenotypic and chemotypic characterization
Three of the obtained strains were phenotypically and chemotypically characterized with the same set of investigations (Pitt et al. 2024) and the same methods (Pitt et al. 2022) as reported previously. In brief, potential anaerobic growth, the temperature range of growth, and tolerance for NaCl were tested in each case on NSY agar plates inoculated with a well-growing liquid culture. For testing anaerobic growth, an anaerobic jar (oxygen removed with crystalline silicic acid) and in addition to standard NSY plates, NSY plates supplemented with 2 g l^−1^ NaNO_3_ were used to specifically test for nitrate respiration. Temperatures were tested starting at 5 °C and increased if the strains showed growth. Close to the supposed tolerated temperature, we used 1 °C steps until the strains showed no growth. The same procedure served for testing NaCl tolerance by using steps of 0.1% w/v. For testing the motility of the strains, soft agar plates (0.1 g l^−1^ K_2_HPO_4_, 1 g l^−1^ yeast extract, and 2.0 g l^−1^ agar) were inoculated with a drop of culture and monitored for fourteen days. Spreading over the whole agar plate within 2 weeks was regarded as a positive result. Cell shapes and metrics were determined with an epifluorescence microscope (UV filter) after fixing liquid cultures with 2% paraformaldehyde and staining by 4′,6-diamidino-2-phenylindole (DAPI). The composition of the fatty acids, polar lipids, and respiratory quinones were determined. For these purposes, the biomass of liquid cultures (NSY medium, room temperature, 3-day incubation) was harvested by centrifugation. After saponification and methylation, the fatty acid content was analyzed by gas chromatography coupled to a flame ionization detector as described previously (Sasser 1990). The identity of fatty acid species was determined by gas chromatography/mass spectrometry (GC/MS) (Vieira et al. 2021). Double bond-containing species were further resolved by derivatization to dimethyl disulfide adducts (Moss and Lambert-Fair 1989) and subsequent GC/MS analysis. The polar lipids were extracted and analyzed by thin-layer chromatography according to the methods of Tindall (Tindall 1990a, 1990b), which was based on the description by Bligh and Dyer (Bligh and Dyer 1959). Total lipids were detected by spraying the plates with dodecamolybdophosphoric acid (Dmp). Specific functional groups were visualized with α-naphthol, ninhydrin, and molybdenum blue. Respiratory quinones were extracted via solid-phase extraction and analyzed by reversed-phase HPLC coupled to a diode array dector as described previously (Vieira et al. 2021).
Results and discussion
Habitats
An overview of the restoration area and the location of the sampling sites is given in Fig. 1, and all data concerning the sampled habitats are shown in Table 1. The site at the river Salzach was characterized by electrical conductivity values between approximately 200 μS/cm and 350 μS/cm and pH values between 7 and 8.5; the water temperatures varied from 5.8 °C in winter to 16.4 °C during the summer sampling (Table 1). The data of the nearby creek Reitbach showed similar magnitudes but higher fluctuations (Table 1). The fluctuations were even higher at the two sampling sites with standing waters, so the lake Ausee and the newly created small pond. They reached high temperatures of about 25 °C in summer and low temperatures of around 4 °C in winter. Probably due to high primary productivity, the pH value raised in summer in the case of the Ausee to almost 9. The saturation of oxygen was in all four habitats during the sampling period between 80 and 100%; therefore, enough oxygen for aerobic bacteria was available in the upper water layers (sampling at 0.1 m depth). Table 1. Characteristics of the four habitats of the investigated area and taxonomic assignment of the isolated strains. Messurements were performed in 0.1-m depth. 1, Salzach; 2, newly created pond; 3, Ausee; 4, Reitbach1234Habitat typeRiverSmall pondLakeCreekGeographic coordinates47.914518 N12.954952 E47.923345 N12.962754 E47.920563 N12.959348 E47.914978 N12.956025 EWidth or area158 m0.16 ha9.99 ha9 mSampling 12.07.2023 pH8.58.98.1n.d.* Electrical conductivity214 μS/cm174 μS/cm370 μS/cmn.d.* Water temperature16.4 °C25.9 °C25.3 °Cn.d.* Oxygen9.1 mg/L, 96.3%7.8 mg/L, 99.6%7.7 mg/L, 97.8%n.d.* Isolated strains belong toA. saliriviA. ecclesiiriviA. antheringensisNo cultures obtainedNo cultures obtainedNo culturing experimentSampling 8.11.2023 pH7.26.76.88.1 Electrical conductivity297 μS/cm265 μS/cm350 μS/cm455 μS/cm Water temperature9.3 °C8.5 °C10.4 °C8.6 °C Oxygen9.15 mg/L, 83.5%9.66 mg/L, 86.1%8.8 mg/L, 81.7%9.33 mg/L, 84.4% Isolated strains belong toA. antheringensisA. novilacunae**A. antheringensisA. novilacunaeA. ecclesiiriviA. regiilacusA. aurantiipilulaA. sp.A. antheringensis**A. nivalisilvaeSampling 31.1.2024 pH7.68.17.48.5 Electrical conductivity348 μS/cm296 μS/cm360 μS/cm468 μS/cm Water temperature5.8 °C4.3 °C4.8 °C4.1 °C Oxygen10.4 mg/L, 86%12.7 mg/L, 102.8%9.5 mg/L, 80.1%11.5 mg/L, 90.3% Isolated strains belong toNo culturing experimentNo culturing experimentNo culturing experimentNo culturing experimentSampling 16.04.2024 pH7.47.97.86.7 Electrical conductivity196 μS/cm352 μS/cm350 μS/cm233 μS/cm Water temperature15 °C14.3 °C14.2 °C9.6 °C Oxygen n.d9.0 mg/L, 91.9%10.3 mg/L, 104.8%10.5 mg/L, 97.2% Isolated strains belong toNo cultures obtainedNo cultures obtainedNo cultures obtainedNo cultures obtained^*^Additional sampling 03.09.2024 pH 7.9, conductivity 406 μS/cm, temperature 19.4 °C, oxygen 83.9%
Isolated strains and metagenome of the lake Ausee
The water samples taken in the summer and autumn of 2023 and the spring of 2024 were used to get bacterial strains belonging to the genus Aquirufa (Table 1). Interestingly, the sampling in summer resulted only in the case of the river Salzach in Aquirufa isolates. The obtained strains belonged to two known species, and one of them represented a new species (Table 1). While not a single Aquirufa isolate was received from the samplings in spring, we got pure cultures from all four water samples in autumn (Table 1). Numerous strains originated from the lake Ausee, they belonged to six different species, including two so far undescribed ones (Table 1). For further analyses aiming to describe new Aquirufa species, we selected strain 1-SAACH-A3^T^ originating from the river Salzach, and 2-BAHN-186B^T^ and AUSEE-184A6 originating from the newly created pond and the lake Ausee, respectively.
The analyses of the metagenome sequence of the autumn sample of the Ausee could not confirm the findings of the cultivation experiment. Mapping of the obtained metagenomic reads on Aquirufa genomes resulted only in the detection of A. antheringensis (coverage breadth 60.3% of the genome, 3.1 reads/covered position). The metagenome assembly and binning resulted in a metagenome-assembled genome (MAG) with 0.7 Mbp (276 contigs) with 28.2% completeness, 0.86% contamination, but 100% heterogeneity. The taxonomic assignment revealed that it should belong to unknown Aquirufa species. Calculation of gANI value with Aquirufa strains resulted in 89.7% for the type strain of A. antheringensis as the highest value, with a coverage of 68.6%. Thus, the MAG could not be assigned to a previously described species or a new species discovered through the cultivation experiments, likely it consisted of small parts of two or more unknown Aquirufa species. Finally, BLAST comparisons with gyrB gene sequences of genome-sequenced Aquirufa strains against the assembled metagenomic contigs resulted in the detection of A. antheringensis (identity 97%) and strain 2-AUSEE-184A6 (identity 99% and 100%, respectively). All these results suggested that Aquirufa populations were not abundant enough in the lake Ausee sample to get precise information about the species composition through metagenome sequencing.
Strains representing new species
Phenotypic and chemotypic characteristics
The phenotypic and chemotypic characteristics of the three strains, 1-SAACH-A3^T^, 2-BAHN-186B^T^, and 2-AUSEE-184A6 are shown in Table 2, the fatty acid compositions in Table S2, and the patterns of the polar lipids in Fig. S2. All characteristics of these strains resembled the characteristics of the related type strains, only slight differences concerning the respiratory quinones, the fatty acids, and polar lipids could be observed (Table 2). Table 2. Features of the new strains and the type strains of the nearest related species1234****567Mean cell length (μm)1.21.71.51.21.41.71.2Mean cell width (μm)0.40.50.30.40.60.60.5Temperature range for growth (°C)5–32 (w)5–345–30 (w)5–32 (w)5–32 (w)5–32 (w)5–31 (w)NaCl tolerance (%, w/v)0–0.20–0.40–0.2 (w)0–0.10–0.10–0.3 (w)0–0.1 (w)Respiratory quinones (% of the total content): Menaquinone-796.7100.0 ≥ 99.094.795.5 ≥ 99.099.4 Menaquinone-63.1- < 1.03.43.2 < 1.0- Menaquinone-80.2--1.91.3-0.6Major fatty acids (% of the total content): C_16:1_ω5c9.513.96.710.18.111.97.0 C_16:1_ω7c23.121.021.012.16.924.712.0 iso-C_15:0_21.824.522.734.948.020.341.4 anteiso-C_15:0_8.88.48.311.313.25.615.5 iso-C_15:0_3-OH10.89.38.39.44.713.44.5Polar lipids:* Unidentified phospholipids1-11--- Unidentified aminolipid--1---- Unidentified aminophospholipids3333223 Unidentified glycolipids2--32-- Unidentified polar lipids42233421, 1-SAACH-A3^T^; 2, A. ecclesiirivi 50A-KIRBA^T^; 3, A. beregesia 50C-KIRBA^T^; 4, 2-BAHN-186B^T^; 5, 2-AUSEE-184A6; 6, A. antheringensis 30S-ANTBAC^T^;** 7**, A lenticrescens 9H-EGSE^T^All strains had in common: cell morphology: rods, pigmentation of colonies: red, pigmentation in liquid medium: red-orange, anaerobic growth: -; gliding on soft agar: + ; identified polar lipid: phosphatidylethanolamine. Only the major fatty acids (≥ 10% for at least one strain) are listed, the whole fatty acid composition of the new strains is shown in Table S2. *, and/or iso-C_15:0_2-OH; − , negative; + , positive; w, weakAll data were elevated in the same laboratories under the same conditions, data from columns 2, 3, 6, and 7 were published earlier (Pitt et al. 2019, 2020, 2022, 2024)
Phylogenetic position
Phylogenetic reconstructions with 16S rRNA gene sequences placed strain 1-SAACH-A3^T^ on the A. nivalisilvae branch of the genus Aquirufa; 2-BAHN-186B^T^ and 2-AUSEE-184A6 were situated on the A. antheringensis branch (Fig. S3). BLAST searches with the NCBI database (Johnson et al. 2008) revealed 16S rRNA gene sequence similarity values for strain 1-SAACH-A3^T^ of 99.9% compared with nearly all species of the branch (A. rosea 99.5%). The two other new strains had identical 16S rRNA gene sequences, the highest similarity values amounted to 99.9% and 99.8% for comparison with A. lenticrescens and A. antheringensis, respectively*.* The genome-based phylogenetic tree calculated by using amino acid sequences of 658 core genes (Fig. 2) showed a more differentiated insight into the phylogeny of the new strains. While the allocation of the new strains to the two Aquirufa branches was reflected as well, 1-SAACH-A3^T^ was very close to A. ecclesiirivi, while 2-BAHN-186B^T^ and 2-AUSEE-184A6 were on a separated branch beside Aquirufa lenticrescens and Aquirufa antheringensis (Fig. 2). Overall, the genome-based tree revealed the phylogenetic distances between the regarded strains, which could not be recognized in the 16S rRNA gene-based tree.Fig. 2. Phylogenetic reconstruction of the new strains, all Aquirufa type strains and type strains of closely related genera. Shown is a midpoint-rooted RaxML tree based on the amino acid sequences of 658 core genes. Bootstrap values for all nodes 100%
Genomic characteristics
The authenticity of all assembled genomes could be confirmed by comparison of the 16S rRNA gene Sanger sequences with the genome sequences. The genome of strain 1-SAACH-A3^T^ consisted of 56 contigs, had a nucleotide coverage of 403 × , a size of 3.2 Mbp and a G + C content of 38.4 mol%. The strain fit regarding the genome size and a G + C content of 38.4% very well to the related species of the A. antheringensis branch (Table 3). Like the type strains of A. ecclesiirivi and A. beregesia, the genome of 1-SAACH-A3^T^ included an operon for the assimilatory usage of nitrate and nitrite including genes predicted for nitrate reductase, nitrite reductase, a nitrate/nitrite MFS transporter, and rubredoxin. The genome contained also an operon encoding genes for the reduction of nitrous oxide to molecular nitrogen including genes predicted for nitrous oxide reductase and cytochrome c551/c552 (Table 3). The assembled genome of strain 2-BAHN-186B^T^ consisted of 6 contigs, had a nucleotide coverage of 762 × , a size of 2.4 Mbp and a G + C content of 42.4 mol%, of strain 2-AUSEE-184A6 of 7 contigs, coverage 1423 × , a size of 2.4 Mbp and a G + C content of 42.2 mol%. They matched very well with the species of the A. antheringensis branch regarding the genome size, G + C content, typical high coding density, and the number of protein-coding genes (Table 3). Like the type strains of A. antheringensis and A. lenticrescens, the genomes of the two strains encoded genes predicted for the light-harvesting rhodopsin system (Table 3). Table 3. Genomic comparison of the new strains and type strains of related species1234567IMG/MER ID number807,111763282887944628163321248103260213809916898928163321202857132225Genome size (Mbp)3.23.13.22.42.42.52.5DNA G + C (mol%)38.438.438.542.442.242.642.2Coding density92.592.392.295.095.194.895.0Protein coding genes2742266327432158216822242218gANI with 1-SAACH-A3^T^ (%)10093.6588.2875.4375.4175.4175.45Corresponding average AF (%)10090.886.4141.2441.040.1839.92gANI with 2-BAHN-186B^T^ (%)75.4375.3775.3810095.7682.9982.97Corresponding average AF (%)32.232.3931.1910093.8285.4684.8dDDH with 1-SAACH-A3^T^ (d_4_, %)10051.434.718.418.319.018.5dDDH with 2-BAHN-186B^T^ (d_4_, %)18.418.118.210065.560.560.7Genes putatively encoding for:Bacteriorhodopsin COG5524--- + + + + Beta-carotene 15,15’-dioxygenase EC:1.13.11.63--- + + + + Nitrous oxide reductase EC:1.7.2.4 + + + ----Nitrate reductase (assimilatory) EC:1.7.7.2 + + + -- + -Nitrite reductase (assimilatory) EC:1.7.1.15 + + + -- + -Nitrate/nitrite transporter COG2223 + + + -- + -1, 1-SAACH-A3^T^; 2, A. ecclesiirivi 50A-KIRBA^T^; 3, A. beregesia 50C-KIRBA^T^; 4, 2-BAHN-186B^T^; 5, 2-AUSEE-184A6; 6, A. antheringensis 30S-ANTBAC^T^;** 7**, A lenticrescens 9H-EGSE^T^
Ecology and distribution
Like the related type strains of the A. nivalisilvae branch (Table 3), 1-SAACH-A3^T^ showed properties which could be interpreted as a combination of pelagic lifestyle and association with particles or surfaces (Chiriac et al. 2023) such as small cell sizes, intermediate genome sizes, and genes for uptake of nitrate or utilization of nitrous oxide, which occurs at the aerobic/anaerobic layer. Interestingly, neither further cultivation efforts in Austria (Salzburg and Upper Austria) (unpublished data) nor the analysis of 123 publicly available metagenomes indicated the occurrence of the new species represented by 1-SAACH-A3^T^ in other freshwaters. So, it seemed that the strain represents a rare species.
In contrast, strains 2-BAHN-186B^T^ and 2-AUSEE-184A6 showed characteristic properties of freshwater bacteria with a pelagic lifestyle (Chiriac et al. 2023) such as small cell sizes, comparatively low genome sizes, high coding densities of the genomes, and light-harvesting systems, like the related type strains of the A. antheringensis branch (Table 3). Culturing experiments with freshwater samples derived from various sites in Austria (Salzburg and Upper Austria) resulted in no further strains belonging to the species represented by 2-BAHN-186B^T^ and 2-AUSEE-184A6 (unpublished data). Nevertheless, the investigation of 123 publicly available metagenomes from freshwater habitats resulted in 15 cases in detections of the new species represented by the two strains (Table S3). While evidence for the new species was exclusively found in rivers, most of these habitats were located in Asia, the River Geum, The Republic of Korea, and the Jinsha and Yangtze Rivers in China. Two detections were also made in water samples of the Columbia River Estuary, USA, and the Torrens River, Australia (Table S3). It was difficult to quantify the relative abundances of the new species based on the metagenomic sequences, especially because freshwater bacteria have a wide range of genome sizes, which can lead to an over- or underestimation. Since the new species has an average genome size considering planktonic freshwater bacteria, the percentage of metagenomic reads mapping on the genome of strain BAHN-186B^T^ (Table S3) could give insights into the relative abundances. According to these data the highest abundances of the new species were detected in water samples from the Yangtze River (SRR9924798 and SRR9924799, Table S3) with 0.188% and 0.106% mapped reads, respectively. Both habitats were situated in the subtropical climate zone.
The data of the metagenome analyses and the data of the home habitats of the two strains representing the new species suggested that it occurs in standing and running freshwaters as well, maybe in populations with adaptations to the habitat conditions through different genotypes.
Conclusions and evidence for two new species
Concerning the culturing experiments, the greatest biodiversity of Aquirufa species was found in the lake Ausee with six distinct species, while water samples from the river Salzach, the creek Reitbach, and the small recently established pond resulted only in cultures of three, two, and one species, respectively. It is necessary to consider that certainly not all of the present species could be detected by culturing experiments. Since metagenomic sequencing of the lake Ausee sample was not appropriate to get insights into the biodiversity of the genus Aquirufa, further methods are needed. Nevertheless, the results showed some interesting points. First, it seemed that the lake Ausee served as a habitat for a larger number of Aquirufa species; maybe, it acts as a reservoir for the populations to colonize the surrounding water bodies. Second, the occurrence of Aquirufa strains in the upper water layer seems to undergo seasonal fluctuations, with a peak in autumn.
Of special interest were the three new strains, which represent new species. Table 3 indicates the overall genome-related indexes gANI and dDDH of the new strains and the nearest related type strains of Aquirufa species. In the case of 1-SAACH-A3^T^, all gANI values were below the proposed threshold of 95% (Konstantinidis et al. 2006; Jain et al. 2018) but in the case of the type strain of Aquirufa ecclesiirivi with 93.65%, close to the threshold value. Calculations of dDDH values with the same genome set (Table 3) resulted in a maximum value of 51.4% and were more significantly below the used threshold of 70% (Chun et al. 2018). The calculated gANI value comparing strains 2-BAHN-186B^T^ and AUSEE-184A6 of 95.76% was also unusual because it was very close but above the proposed threshold. Comparisons with the closest related type strains of Aquirufa species revealed gANI values around 83%, which indicated clearly that they represent at least one new species. Determination of the dDDH value comparing 2-BAHN-186B^T^ and AUSEE-184A6 resulted in 65.5%, a value slightly below the proposed threshold of species separation. So, more evidence was needed for the assumption that strain 1-SAACH-A3^T^ represented a new species and that strains 2-BAHN-186B^T^ and AUSEE-184A6 belonged to the same species. Figure 3 shows BLAST comparisons of the latter two strains and the pair 1-SAACH-A3^T^/50A-KIRBA^T^ (type strain of A. ecclesiirivi). The figure displayed in the case of 1-SAACH-A3^T^/50A-KIRBA^T^ (Fig. 3, left side) regions with comparatively low identity values for nucleotides and for amino acids, the comparison of strains 2-BAHN-186B^T^ and AUSEE-184A6 yielded a more uniform pattern (Fig. 3, right side). Since calculating gANI values is the most used and accepted method for species demarcation, we focused on deeper insights into the prevalence. As strain 1-SAACH-A3^T^ belongs to the A. nivalisilvae branch of the genus Aquirufa and 2-BAHN-186B^T^ and AUSEE-184A6 to the A. antheringensis branch, we checked if there are any differences concerning the intra-species gANI values and corresponding AF values between the two groups. Figure 4 shows a plot of 107 intra-species gANI/AF value pairs derived from genome sequences of strains belonging to Aquirufa species (Table S1); in addition, strains 2-BAHN-186B^T^ compared with AUSEE-184A6 (bright green dot) and 1-SAACH-A3^T^ compared with 50A-KIRBA^T^ (orange dot). The figure showed an interesting pattern. On the one hand, strains belonging to the A. antheringensis branch were characterized by comparably low intra-species gANI values and strains of the A. nivalisilvae branch by higher gANI values (Fig. 4). If we assume that strain 1-SAACH-A3^T^ represents a new species and strains, 2-BAHN-186B^T^ and AUSEE-184A6 represent together a second new species; the three new strains fit very well into the picture. A model by Hoetzinger et al. (2024) tries to explain different scales in intra-species ANI values. Accordingly, species with comparatively low ANI values consist of bigger and more stable populations which are hardly connected by gene flow. On the other hand, species with higher ANI values consist of small fluctuating populations with high gene flow (Hoetzinger et al. 2024). The observed higher horizontal gene transfer for strain 1-SAACH-A3^T^ compared with strain 2-BAHN-186B^T^ (Fig. 3) fit the model well. While 1-SAACH-A3^T^ was the only received strain belonging to the new species, several strains which could be assigned to the other new species by sequencing of the gyrB gene were cultured from the water samples of the lake Ausee and the newly created pond (data not shown). This could be a hint that the population sizes were here larger. In addition, the genome-based phylogenetic tree (Fig. 2) confirmed the assumption that the three new strains represent two new species. The branch lengths, representing the phylogenetic distance between the strains 2-BAHN-186B^T^ and 2-AUSEE-184A6, were comparably small; however, the phylogenetic distance between 1-SAACH-A3^T^ and the nearest relative A. ecclesiirivi was in the same range as between other Aquirufa species or species of the genus Flectobacillus (Fig. 2). As mentioned above, further investigations are necessary to explore the biodiversity and distribution of the genus Aquirufa more precisely. In this study, we propose the establishment of two new species, Aquirufa salirivi sp. nov. with 1-SAACH-A3^T^ as type strain and Aquirufa novilacunae sp. nov. with 2-BAHN-186B^T^ as type strain.Fig. 3. Visualization of the genomes of strains 1-SAACH-A3^T^ and 2-BAHN-186B^T^. Shown is the G + C-content (peaks indicate values higher or lower than the average G + C content), score of putative horizontal gene transfer and comparison with strain 50A-KIRBA.^T^ and strain 2-AUSEE-184-A6, respectively as BLAST results for nucleotides (inner blue circle) and BLAST results for amino acids (outer blue circle)Fig. 4. Intra-species gANI (whole genome average nucleotide identity) values and alignment fraction values of strains of the genus Aquirufa (circle points) and inter-species values of the new strain 1-SAACH-A3^T^ compared with its closest known relative A. ecclesiirivi strain 50A-KIRBA.^T^ (square point). The colors of the points reflect the assignment to a particular species and to one of the clusters A. antheringensis branch and A. nivalisilvae branch, respectively (see legend)
Description of Aquirufa salirivi sp. nov.
Aquirufa salirivi (sa.li.ri’vi. L. masc. n. sal, salt; L. masc. n. rivus, river; N.L. gen. n. salirivi, of a salt river, referring to the river Salzach, named after transporting salt on it).
Cells of the type strain form rods with an approximate size of 1.2 × 0.4 μm. Liquid cultures have a red–orange coloring, and colonies on agar plates are red-pigmented. Cultures show motility on soft agar plates and grow at 5–32 °C and in 0–0.2% (w/v) NaCl. Major fatty acids are C_16:1_ω7c, iso-C_15:0_, and iso-C_15:0_3-OH. Polar lipids are phosphatidylethanolamine, unidentified phospholipids, unidentified aminophospholipids, unidentified glycolipids, and further unidentified lipids. Menaquinone-7 occurs as a major respiratory quinone, small amounts of menaquinone-6 and traces of menaquinone-8 are present. The genomic DNA of the type strain has 3.2 Mbp and a G + C content of 38.4 mol%.
The type strain is 1-SAACH-A3^T^ (= DSM 117800^ T^ = JCM 37097^ T^), isolated from water of the river Salzach (Salzburg, Austria). Sequence accession numbers: 16S rRNA gene PQ394666, whole genome JBEWZH000000000.
Description of Aquirufa novilacunae sp. nov.
Aquirufa novilacunae (no.vi.la.cu’nae. L. masc. adj. novus, new; L. fem. n. lacuna, pond; N.L. gen. n. novilacunae, of a new pond).
Cells form rods with an approximate size of 1.3 × 0.5 μm. Liquid cultures have a red–orange coloring, and colonies on agar plates are red-pigmented. Cultures show motility on soft agar plates and grow at 5–32 °C and in 0–0.1% (w/v) NaCl. Major fatty acids are iso-C_15:0_, C_16:1_ω7c, anteiso-C_15:0_, C_16:1_ω5c, and C_16:1_ω7c. Polar lipids are phosphatidylethanolamine, unidentified phospholipids, unidentified aminophospholipids, unidentified glycolipids, and further unidentified lipids. Menaquinone-7 occurs as a major respiratory quinone, small amounts of menaquinone-6 and menaquinone-8 are present. The genomic DNA of the type strain has 2.4 Mbp and a G + C content of 42.4 mol%.
The type strain is 2-BAHN-186B^T^ (= DSM 118143^ T^ = JCM 37099^ T^), isolated from the water of a small pond in Weitwörth (Salzburg, Austria). Sequence accession numbers: 16S rRNA gene PQ397515, whole genome JBEWZF000000000. Strain AUSEE-184A6 (= DSM 117801 = JCM 37098), isolated from water of the lake Ausee in Weitwörth (Salzburg, Austria) belongs also to the species. Sequence accession numbers: 16S rRNA gene PQ394667 accession number whole-genome sequence: JBEWZG000000000.
Supplementary Information
Below is the link to the electronic supplementary material.Supplementary file1 (PDF 633 KB)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1García-López M, Meier-Kolthoff JP, Tindall BJ et al (2019) Analysis of 1,000 type-strain genomes improves taxonomic classification of Bacteroidetes. Front Microbiol 10. 10.3389/fmicb.2019.0208310.3389/fmicb.2019.02083 PMC 676799431608019 · doi ↗ · pubmed ↗
- 2Hoetzinger M, Hahn MW, Andersson LY et al (2024) Geographic population structure and distinct intra-population dynamics of globally abundant freshwater bacteria. The ISME Journal 18. 10.1093/ismejo/wrae 11310.1093/ismejo/wrae 113PMC 1128372038959851 · doi ↗ · pubmed ↗
- 3Land-Salzburg (2021) LIFE Project Salzachauen Riparian forest renaturation & visitor guidance. https://www.salzachauen.at/index.php?file=life_salzachauen_expert_publication_en_web.pdf. Accessed 07.02.2025
- 4Lu H, Chen L, Huang L (2024) Arcicella gelida sp. nov. and Arcicella lustrica sp. nov., isolated from streams in China and re-examining the taxonomic status of all the genera within the families Spirosomataceae and Cytophagaceae. Int J Syst Evol Microbiol 74(4). 10.1099/ijsem.0.00634510.1099/ijsem.0.00634538629951 · doi ↗ · pubmed ↗
- 5R Core Team (2020) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/. Accessed 07.02.2025
- 6R Studio Team (2019) R Studio: Integrated development for R. R Studio, Inc., Boston, MA. http://www.rstudio.com/. Accessed 07.02.2025
- 7Schoch CL, Ciufo S, Domrachev M et al (2020) NCBI Taxonomy: a comprehensive update on curation, resources and tools. Database (Oxford) 2020. 10.1093/database/baaa 06210.1093/database/baaa 062PMC 740818732761142 · doi ↗ · pubmed ↗
- 8Vieira S, Huber KJ, Neumann-Schaal M et al (2021) Usitatibacter rugosus gen. nov., sp. nov. and Usitatibacter palustris sp. nov., novel members of Usitatibacteraceae fam. nov. within the order Nitrosomonadales isolated from soil. Int J Syst Evol Microbiol 71. 10.1099/ijsem.0.00463110.1099/ijsem.0.00463133433313 · doi ↗ · pubmed ↗