Turbo‐charging crop improvement: harnessing multiplex editing for polygenic trait engineering and beyond
Kangquan Yin, Chung‐Jui Tsai

TL;DR
Multiplex CRISPR editing is transforming crop improvement by enabling precise, simultaneous modification of multiple genes and traits.
Contribution
The paper reviews recent innovations in multiplex CRISPR editing and their impact on polygenic trait engineering in crops.
Findings
Multiplex editing enables efficient trait stacking and de novo domestication in plants.
High-throughput sequencing improves detection of complex editing outcomes like structural rearrangements.
Computational tools and synthetic biology-compatible workflows are advancing scalability and precision.
Abstract
Multiplex CRISPR editing has emerged as a transformative platform for plant genome engineering, enabling the simultaneous targeting of multiple genes, regulatory elements, or chromosomal regions. This approach is effective for dissecting gene family functions, addressing genetic redundancy, engineering polygenic traits, and accelerating trait stacking and de novo domestication. Its applications now extend beyond standard gene knockouts to include epigenetic and transcriptional regulation, chromosomal engineering, and transgene‐free editing. These capabilities are advancing crop improvement not only in annual species but also in more complex systems such as polyploids, undomesticated wild relatives, and species with long generation times. At the same time, multiplex editing presents technical challenges, including complex construct design and the need for robust, scalable mutation…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
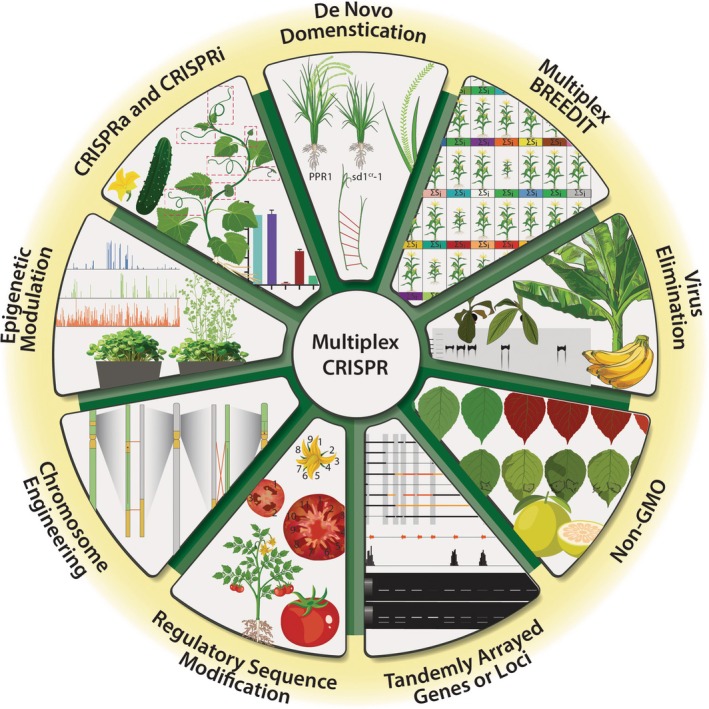 Figure 1
Figure 1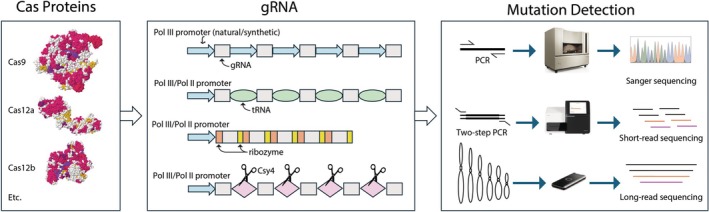 Figure 2
Figure 2| Species | Trait(s) | Target # | Cas protein | gRNA # | gRNA architecture | Efficiency | Detection | Cross | Transgene‐free | References |
|---|---|---|---|---|---|---|---|---|---|---|
| Knockout | ||||||||||
|
| Cell wall | 3 genes | Cas9 | 3–4 | Individual Pol III | Not provided | Sanger | Selfing | Yes | Zhang et al. ( |
|
| n/a | 12 genes | Cas9 | 24 | Individual Pol III | 0–94% | Sanger | Selfing | Yes (some) | Stuttmann et al. ( |
|
| Flowering | 3 genes | Cas12a | 6 | cRNA array (Pol II) | 0–25% | Sanger, WGS | Selfing | Unknown | Jordan et al. ( |
|
| Cell wall | 3 genes | Cas9 | 4–5 | Individual Pol III, tRNA (Pol III) | Not provided | Sanger | Selfing | Unknown | Zhang et al. ( |
|
| Organ ablation | repeats | Cas9 | 2 | Individual Pol III | Up to 89% | Amp‐seq | Selfing | Unknown | Schindele et al. ( |
|
| Growth | 8 genes | Cas9 | 2–8 | Individual Pol III, tRNA (Pol III) | 0–93% | Amp‐seq | Selfing, intercross | Yes | Angulo et al. ( |
|
| Cell ablation | repeats | Cas9 | 1–2 | Individual Pol III | Not provided | Fluorescence imaging | Selfing | Unknown | Gehrke et al. ( |
|
| Disease resistance | 1 gene, 1 promoter | Cas12a, nCas9‐APOBEC1 | 2 | tRNA (Pol III), ribozyme (Pol III) | 2% transgene‐free | Sanger, WGS | Yes | Huang et al. ( | |
|
| Disease resistance | 1 gene | Cas12a | 3 | Direct synthesis | 100% | Sanger, WGS | Yes | Su et al. ( | |
|
| Disease resistance | 3 genes | Cas9 | 3 | tRNA (Pol II) | 0–100% | Amp‐seq | Selfing | Yes | Ma, Yang, et al. ( |
|
| Nodulation | >102 genes | Cas9 | 1 | Individual Pol III | 49% overall | Sanger | Selfing | Yes (some) | Bai et al. ( |
|
| Oligosaccharides | 2 genes | Cas9 | 4 | tRNA (Pol III) | 33–83% | Sanger | Selfing | Yes | Cao et al. ( |
|
| Resistant starch | 7 genes | Cas9 | 7 | Individual Pol III | 0–74% | PCR | Selfing | Unknown | Yang et al. ( |
|
| Disease resistance, male sterility | 3 genes | Cas9 | 3 | Individual Pol III | Not provided | Sanger | Selfing | Yes | Li et al. ( |
|
| Resistant starch | 4 genes | Cas9 | 4 | tRNA (Pol III) | 25–88% | Amp‐seq | Selfing | No | Biswas et al. ( |
|
| Growth | 12 genes | Cas9 | 6 or 12 | Individual Pol III | 68–100% | Amp‐seq, WGS | Selfing, intercross | Yes | Wei et al. ( |
|
| Flowering | 2 genes | nCas9‐APOBEC3A | 2 | Individual Pol III | 2% transgene‐free, 7% biallelic CEN1 | Sanger, WGS | Yes | Wu et al. ( | |
|
| Lignin | 2 genes | Cas9 | 1 (2 sets) | Individual Pol III | 0–62% | Sanger | No | de Vries et al. ( | |
|
| Trichome | 3 genes (8 alleles) | Cas9 | 1 | Individual Pol III | 97–100% | Amp‐seq | No | Bewg et al. ( | |
|
| Reproduction | 2–6 genes | Cas9 | 1–3 | Individual Pol III | 100% | Amp‐seq | No | Ortega et al. ( | |
|
| Lignin | 3 genes | nCas9‐APOBEC3A | 2 | Individual Pol III | 7% transgene‐free, 0% biallelic CCoAOMT1 | Sanger, WGS | Yes | Hoengenaert et al. ( | |
|
| Lignin | 10 genes | Cas9 | 3–6 (7 sets) | tRNA (Pol III) | 0–100% | Sanger, Amp‐seq | No | Sulis et al. ( | |
|
| Multiple traits | 8 genes | Cas12a, nCas9‐APOBEC3A | 2 | tRNA (Pol III), ribozyme (Pol II) | 8–42% transgene‐free | Sanger, WGS | Yes | Huang et al. ( | |
|
| Fruit color | 3 genes | Cas9 | 6 | tRNA (Pol III) | 8–92% | Sanger | BC, selfing | Yes | Yang et al. ( |
|
| Yield, drought tolerance | 48 genes | Cas9 | 12 (4 sets) | Individual Pol III | 73% overall | Amp‐seq | Selfing, intercross | Yes (some) | Lorenzo et al. ( |
| Editing of tandemly arrayed genes | ||||||||||
|
| Disease resistance | 4 TAGs | Cas9 | 2 | Individual Pol III | Not provided | Sanger | Selfing | No | Shen et al. ( |
|
| Reproduction | 7 TAGs | Cas9 | 2 or 6 | Individual Pol III | Not provided | Sanger | Selfing | Yes | Zhong et al. ( |
|
| n/a | Mulittags of 2–6 | Cas9 | 2 (multisets) | Individual Pol III | 0–78% TAG deletions | PCR, Sanger | Selfing | Unknown | Liu et al. ( |
|
| Disease resistance | 2 genes | Cas9 | 4 | Individual Pol III | Not provided | Sanger | Unknown | Shen et al. ( | |
|
| n/a | 7 TAGs | Cas9 | 1 | Individual Pol III | 98–100% | PCR, Amp‐seq, Capture‐seq | No | Chen et al. ( | |
| Polyploid engineering | ||||||||||
|
| Height | 3 genes | Cas9 | 1 | Individual Pol III | 40% | Sanger | No | Ye et al. ( | |
|
| Virus resistance | 3 genes | Cas9 | 3 | Individual Pol III | 70–85% | Sanger | No | Tripathi et al. ( | |
|
| Herbicide tolerance | 2 genes | nCas9‐APOBEC1 | 1–2 | Individual Pol III | 45–53% (1–3% transgene free) | Sanger, WGS | Yes | Van den Broeck et al. ( | |
|
| Therapeutic proteins | 6 genes | Cas9 | 3–7 | tRNA (Pol III) | 0–83% | Sanger | Selfing | Unknown | Jansing et al. ( |
|
| n/a | 9 genes | Cas9 | 10 | Individual Pol III | 96.50% | Sanger | No | Stuttmann et al. ( | |
|
| Domestication | Many | Cas9 | 2–8 (multisets) | Individual Pol III | Not provided | Sanger | No | Yu et al. ( | |
|
| Chlorophyll | 59 alleles | Cas9 | 2 | Individual Pol III | 14–20% | Sanger and Amp‐seq | No | Eid et al. ( | |
|
| Allergen reduction | >17 targets | Cas9 | 7 | tRNA (Pol III) | 21–100% | Sanger | Selfing | Yes | Camerlengo et al. ( |
|
| Resistant starch | 3 genes | Cas9 | 1 (2 sets) | Individual Pol III | 33–67% | Sanger | Selfing | Yes | Li et al. ( |
|
| Multiple traits | 6 genes | Cas9 | 2–5 | tRNA (Pol II) | 18–75% | PCR, Sanger | Selfing, embryo rescue | Yes | Luo et al. ( |
|
| Allergen reduction | >32 targets | Cas9 | 2 or 4 | Individual Pol III | 0–100% (34% overall) | Sanger and Amp‐seq | Selfing | Yes | Sánchez‐León et al. ( |
|
| Allergen reduction | >89 targets | Cas9 | 1 (7 sets) | Csy4 (Pol II) | Not provided | PCR, Amp‐seq, WGS | Selfing | Yes | Yu et al. ( |
| Transcriptional regulation (including combinatorial editing) | ||||||||||
|
| Flowering time | 1 promoter (methylation) | dCas9 | 3 | Individual Pol III | 25% (DNA methylation) | RT‐qPCR, bisulfite PCR sequencing | Selfing | Yes | Ghoshal et al. ( |
|
| Rapid breeding | 1 promoter, 4 genes | Cas9, nCas9‐APOBEC3A | 2 | Individual Pol III | ~3–65% | Amp‐seq | Selfing | Yes | Pan et al. ( |
|
| Flowering | 1 promoter | dCas9 | 2 | Individual Pol III | 80% (activation) | RT‐qPCR | Selfing | No | Zinselmeier et al. ( |
|
| Root flavonoids | 6 promoters | dCas9 | 6–12 | Individual Pol III | Not provided | Fluorescence imaging | Selfing | No | Houbaert et al. ( |
|
| Grain quality | 1 promoter +5’‐UTR | Cas9 | 1–2 | Individual Pol III | Not provided | PCR, Sanger | Selfing | Yes | Zeng et al. ( |
|
| Regeneration | 1 promoter, 2 genes | Cas9 | 2 | tRNA (Pol II) | 37–88% | RT‐qPCR, Amp‐seq | No | Pan et al. ( | |
|
| Yield | 1 promoter + UTRs | Cas9 | 2–4 (39 sets) | Individual Pol III | 19% | PCR, Sanger | Selfing | Yes | Song et al. ( |
|
| Regeneration | 2 promoters, 1 gene | Cas9 | 2 | Individual Pol III | 50–80% | RT‐qPCR, Amp‐seq | No | Pan et al. ( | |
|
| Fruit traits | 2 promoters | Cas9 | 8 | Individual Pol III | Not provided | PCR, Sanger, WGS | BC, selfing | Yes | Rodríguez‐Leal et al. ( |
|
| Productivity | 2 promoters | Cas9 | 8 | Individual Pol III | Not provided | PCR, Sanger | BC, selfing | Yes | Wang et al. ( |
|
| Branching | 1 promoter | Cas9 | 6–8 | Individual Pol III | Not provided | PCR, Sanger, WGS | BC, selfing | Yes | Hendelman et al. ( |
|
| Grain yield | 2 promoters | Cas9 | 9 | Unspecified | Not provided | PCR, Sanger | BC, selfing | Yes | Liu et al. ( |
| Chromosome engineering | ||||||||||
|
| Chr translocation | 2 targets | Cas9 | 2 | Individual Pol III | 0.01–0.05% | PCR, Amp‐seq, FISH | Selfing | Yes | Beying et al. ( |
|
| Chr inversion | 2 targets | Cas9 | 2 | Individual Pol III | 0.50% | PCR, Sanger | Selfing | Unknown | Schmidt et al. ( |
|
| Chr inversion | 2 targets | Cas9 | 2 | Individual Pol III | Not provided | PCR, Sanger, FISH | Selfing | Yes | Rönspies et al. ( |
|
| Chr inversion | Cas9 | 2 | Individual Pol III | 0.06–0.25% | PCR, Sanger, FISH | Selfing | Yes | Khosravi et al. ( | |
|
| Chr inversion | 2 targets | Cas9 | 2 | Direct synthesis | 0.13% | Amp‐seq, Bionano | Selfing | No | Schwartz et al. ( |
- —Georgia Research Alliance10.13039/100008065
- —U.S. Department of Energy10.13039/100000015
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCRISPR and Genetic Engineering · Chromosomal and Genetic Variations · Plant Virus Research Studies
INTRODUCTION
As a disruptive technology, CRISPR‐Cas has made gene editing accessible across all fields of life sciences. Over the past decade, more than 55,000 studies have harnessed CRISPR‐Cas tools to modify genomes, regulate gene expression, and more, in an ever‐growing number of plant species. The system's core components are remarkably simple: a Cas nuclease or its deactivated or modified form, and one or more guide RNAs (gRNAs) that target‐specific genome sites through Watson–Crick base pairing. This simplicity underpins the versatility and adaptability of CRISPR‐Cas to a myriad of applications (Wang & Doudna, 2023) (Boxes 1 and 2).
Box 1Summary of main points
- Multiplex CRISPR editing enables simultaneous manipulation of multiple loci, offering a powerful approach to dissect gene families and overcome genetic redundancy.
- It accelerates polygenic trait engineering and trait stacking, especially in complex systems such as polyploid crops, wild relatives, and perennial species.
- Applications extend beyond gene knockouts to include regulatory sequence editing, epigenetic modulation, chromosomal engineering, and combinatorial genome manipulations.
- Ongoing technical advances in construct design, delivery platforms, and mutation detection methods are expanding the scalability and precision of multiplex editing.
Box 2Open questions
- How can multiplex editing workflows be scaled for high‐throughput applications across diverse plant species and genotypes?
- What strategies can minimize somatic chimerism and enhance the early recovery of homozygous or biallelic edits?
- Can multiplex systems be programmed for spatiotemporal control and combinatorial logic in gene regulation?
- What are the practical limits of multiplex editing in achieving consistent and effective outcomes across target sites?
- How can AI, machine learning, and large language models be leveraged to design, predict, and interpret multiplex editing outcomes?
However, multiplex editing remains relatively underexplored in plant sciences, with fewer than 170 papers adopting this approach at the time of this writing, representing only a small fraction (<3%) of the overall CRISPR‐related plant research published to date. Given the polygenic nature of many agronomic traits and the increasingly sophisticated plant design strategies for combinatorial trait engineering, multiplex editing is essential to fully realize the potential of CRISPR in next‐generation crop improvement (Box 1) (Fagny & Austerlitz, 2021; Gilbertson et al., 2025; Shelake et al., 2022).
Paradoxically, naturally evolved CRISPR systems are adept at multiplex editing in bacteria and archaea. All known CRISPR loci possess arrays of spacers acquired from invading organisms over time, which are deployed through highly effective parallel processing to confer adaptive immunity against subsequent invasions (Mojica et al., 2005). Repurposing CRISPR‐Cas for multiplex editing in eukaryotes, including plants, requires the construction of multiple gRNA expression cassettes and/or artificial CRISPR arrays (Cong et al., 2013; Lowder et al., 2015). However, the repeating scaffolds and other sequence elements in these systems present potential genetic engineering challenges, including construct assembly and genetic stability in both bacterial intermediates (E. coli and Agrobacterium) and the eventual plant hosts (Assaad et al., 1993; Bzymek & Lovett, 2001; Ma, Zhang, et al., 2015; Stuttmann et al., 2021).
In this review, we highlight recent advances in CRISPR multiplex editing, spanning applications from gene family characterization and polygenic trait engineering to higher‐order combinatorial genome modifications and chromosomal engineering. We examine current toolkits, their limitations, and emerging opportunities for innovation. We also discuss the analytical and bioinformatic challenges of detecting and interpreting mutations across multiple target sites, which are inherently more complex than single‐target editing. While we do not cover plant transformation bottlenecks or tissue culture‐independent strategies addressed in recent reviews (Brandizzi et al., 2025; Lee & Wang, 2023; Nasti & Voytas, 2021; Quiroz et al., 2024), we acknowledge their critical role in enabling multiplex applications. Continued exciting breakthroughs are expected in the application of multiplex editing for crop improvement, fueled by rapid advances in CRISPR toolkits and sequencing technologies.
MULTIPLEX EDITING TO OVERCOME GENETIC REDUNDANCY
Gene duplication and gene family characterization
Gene duplications and gene families are pervasive in plant genomes (Flagel & Wendel, 2009). The functional redundancy of paralogous genes or gene family members—whether full, partial, or overlapping—poses a major challenge for the mechanistic dissection of causal genes underlying traits of interest (Iohannes & Jackson, 2023). The precision and versatility of CRISPR editing at the gene, paralog, and family level offer significant improvements over previous gene silencing methods (e.g., antisense, RNAi, microRNA) in differentiating between specific and redundant roles of highly homologous genes.
One such example is powdery mildew resistance, where single‐gene knockouts of the Mildew Resistance Locus O (MLO) confer broad‐spectrum resistance in barley (Hordeum vulgare) and wheat (Triticum aestivum) (Angulo et al., 2023; Büschges et al., 1997; Wang, Naik, et al., 2014), but durable resistance in dicots requires multigene knockouts. In Arabidopsis thaliana, triple mutants (Atmlo2 Atmlo6 Atmlo12) were generated through successive intermutant crosses (Consonni et al., 2006). In cucumber (Cucumis sativus L.), multiplex knockouts of three clade V genes (Csmlo1 Csmlo8 Csmlo11) were necessary to achieve full resistance (Table 1) (Ma, Yang, et al., 2024). These mutants also revealed roles for calcium signaling components in powdery mildew defense, highlighting the mechanistic insights enabled by multiplex approaches (Ma, Yang, et al., 2024). The ability of a single multiplex transformation to generate both single‐ and multigene knockouts in various combinations has greatly accelerated research (Ma, Yang, et al., 2024). In annual species, novel mutations not observed in the T_0_ generation can emerge in sensitized T_1_ populations through selfing or controlled crosses (Table 1), thereby expanding the mutation spectrum without additional rounds of resource‐intensive transformation (Ma, Yang, et al., 2024; Rodríguez‐Leal et al., 2017).
In hybrid poplar (Populus tremula × P. alba INRA 717‐1B4), a single gRNA targeting a conserved sequence among MYB transcription factor genes successfully edited three known paralogs (PtaMYB186, PtaMYB138, and PtaMYB38), along with two additional tandem duplicates in one of the subgenomes that were only identified after sequencing the transformation genotype (Bewg et al., 2022; Zhou et al., 2023). This underscores the importance of accurate genome annotation for interpreting copy number variation and multiplex editing outcomes. Editing all eight alleles resulted in glabrous phenotypes and uncovered a link between triterpene biosynthesis and non‐glandular trichomes in poplar (Bewg et al., 2022). This study also demonstrated that multiplex editing of closely linked genes can induce large genomic deletions (~29–62 Kb). These structural variants are likely underestimated by standard indel‐focused genotyping methods.
In another example, multiplex editing clarified the role of caffeoyl shikimate esterase (CSE) in lignin biosynthesis. While single knockouts of CSE paralogs in poplar had no effect, double mutants showed reduced lignin content and altered composition (de Vries et al., 2021). Mutant characterization further revealed partial redundancy and broader substrate specificity, which were further supported by in vitro enzyme kinetics (de Vries et al., 2021). This work highlights the value of generating both single and multiplex mutant lines to disentangle functional redundancy and specificity.
Multiplex editing is also valuable for gene family investigation in A. thaliana, despite the availability of extensive mutant collections from chemical and insertional mutagenesis (Kim et al., 2006; Krysan et al., 1999). Generating higher‐order mutants by crossing is time‐consuming and often impractical for closely linked genes. Furthermore, traditional mutants frequently harbor secondary mutations (Carrère et al., 2024; Raabe et al., 2024), which complicate data interpretation. CRISPR multiplex editing enables the direct generation of complex mutant combinations in a single generation, streamlining functional analysis. For example, Zhang et al. (2020) used six gRNAs in two constructs to target three GLCAT14 genes encoding β‐glucuronosyltransferases and successfully generated single, double, and triple knockout mutants in the T_1_ generation. Mutant characterization revealed redundant roles of GLCATs in arabinogalactan‐protein (AGP) glycosylation, with pleiotropic effects on seed germination, tip growth, pollen function, and seed coat mucilage (Zhang et al., 2020). Similarly, multiplex editing of five hydroxyproline‐O‐galactosyltransferase genes produced triple and quintuple mutants with additive effects on AGP glycosylation, overall growth, root development, and reproduction (Zhang et al., 2021). These studies demonstrate how multiplex editing accelerates the dissection of gene family function and reveals phenotypic complexity masked by redundancy.
Other recent applications of multiplex editing for investigating functional redundancy include the raffinose synthase family in soybean (Cao et al., 2022) and the growth‐regulating factor (GRF) family in A. thaliana (Angulo et al., 2023). The GRF study represented the cumulative progress of two undergraduate laboratory course sessions and one undergraduate research project, showcasing the power of CRISPR in experiential learning for life science undergraduate education.
Tandemly arrayed genes
Tandemly arrayed genes (TAGs) derived from tandem duplications constitute a substantial portion of plant genomes (Rizzon et al., 2006; Yu et al., 2015). Their high sequence similarity and functional redundancy often necessitate multigene knockouts for effective functional analysis. TAGs can undergo homologous or unequal recombination, leading to either functional diversification or degeneration (Hanada et al., 2008; Otto et al., 2022). Disentangling the functional fates of TAGs is particularly challenging using conventional genetic approaches due to low recombination rates between closely linked loci (Jander & Barth, 2007). Consequently, functional studies of TAGs remain limited.
Multiplex CRISPR editing has enabled functional dissection of TAGs in ways that were previously impractical. In A. thaliana, sequential multiplex transformations targeting the AtLURE1 family revealed that these cysteine‐rich LURE1 peptides promote conspecific pollen tube attraction and reproductive isolation, rather than simply impeding interspecific fertilization (Zhong et al., 2019). Although effective, the need for multiple transformation rounds highlights the limitation of this approach in species with long generation times.
In contrast, a single multiplex transformation in hybrid poplar successfully disrupted all seven Nucleoredoxin1 genes within a ~100 kb TAG cluster using one gRNA (Chen et al., 2023). This generated a wide spectrum of mutations, from small indels to large deletions and complex rearrangements, including translocations, inversions, and multi‐fragment fusions, many of which escaped detection by standard PCR or amplicon sequencing (Chen et al., 2023). Capture sequencing with de novo assembly resolved some of these structural variants (Figure 1, Table 1), underscoring the need for advanced genotyping strategies in TAG editing.
Applications of multiplex CRISPR genome editing in plants.This circular diagram illustrates key applications of multiplex CRISPR in plant systems: (1) CRISPR activation and interference (CRISPRa and CRISPRi) for gene regulation; (2) de novo domestication of wild species; (3) multiplex‐assisted breeding strategies; (4) virus elimination in crops such as banana; (5) development of non‐GMO varieties; (6) editing of tandemly arrayed genes or loci; (7) modification of regulatory sequences; (8) chromosome engineering; and (9) epigenetic modulation. These applications underscore the versatility of multiplex genome editing in advancing both basic research and crop improvement. Sources adapted with permission or under Creative Commons licenses (CC BY/CC BY 4.0).
Similar outcomes have been reported in A. thaliana and rice (Oryza sativa), where multiplex editing of TAGs, encoding phytocytokines, metacaspases, chitinases, and disease resistance proteins led to large deletions and inversions (Table 1) (Liu et al., 2023; Shen et al., 2019; Shen et al., 2022). It should be noted that chromosomal deletions and inversions are not limited to TAGs per se, and can occur between closely linked genes (Liu et al., 2023) or between distant target sites on the same chromosome as directed by gRNAs (Khosravi et al., 2025). While modified PCR assays can distinguish inversions from deletions (Liu et al., 2023), more complex rearrangements remain difficult to detect. These findings highlight both the power and technical challenges of multiplex editing in TAG loci and emphasize the importance of comprehensive mutation profiling.
Polyploid genome engineering
CRISPR genome editing is a game changer for polyploid species with inherently complex genomes. For instance, sugarcane cultivars are polyploid/aneuploid interspecific hybrids of Saccharum officinarum and Saccharum spontaneum, possessing 10–13 sets of the 10 basic chromosomes (Souza et al., 2019). Similarly, woody bamboos encompass over 1500 species of tetraploids or hexaploids with 12 chromosomes (Ma, Liu, et al., 2024). As such, multiplex (multiallele) editing is essential in polyploid species, even for single‐gene targets, to obtain null mutants.
In sugarcane, targeting a chlorophyll biosynthetic gene with two gRNAs resulted in mutations across 49 of 59 alleles, demonstrating the feasibility of multiplex editing to address genetic redundancy in highly polyploid crops (Eid et al., 2021). Similarly, in hexaploid Ma bamboo (Dendrocalamus latiflorus Munro), multiplex editing of a gibberellin‐responsive gene produced null mutants with increased plant height (Ye et al., 2020).
In wheat, multiplex editing has been applied to distinct gene families regulating gluten‐related traits (Table 1). Seven gRNAs were used to edit WTAI (wheat tetrameric α‐amylase/trypsin inhibitor) subunits CM3 and CM16 in an elite durum wheat cultivar to reduce allergens associated with baker's asthma (Camerlengo et al., 2020). In separate studies, multiple gRNAs targeted γ‐ and ω‐gliadin gene clusters in hexaploid bread wheat, achieving 64–97% reductions in gliadin content in homozygous, transgene‐free lines (Sánchez‐León et al., 2024; Yu et al., 2024). Crosses with previously edited α‐gliadin‐deficient lines (Sánchez‐León et al., 2018) further reduced total gluten levels (Sánchez‐León et al., 2024), offering a promising path toward gluten‐free wheat. The complications associated with TAG editing, as discussed in the previous section, are exacerbated in polyploid crops lacking high‐quality reference genomes, underscoring universal challenges for multiplex editing applications.
QUANTITATIVE TRAIT ENGINEERING AND STACKING
Speedy polygenic trait engineering
Plant traits are rarely controlled by single genes, making multiplex gene editing a powerful tool for accelerating trait improvement. In tomato (Solanum lycopersicum), fruit color diversity was engineered by simultaneously editing three genes, PSY1 (phytoene synthase 1), MYB12, and SGR1 (STAYGREEN 1), using two gRNAs per gene (Yang et al., 2023). The green‐fruited psy1 myb12 sgr1 triple mutant was backcrossed with wild type, followed by selfing of transgene‐free F1 plants to generate a segregating population with novel fruit colors in less than a year (Yang et al., 2023).
In cereals, dietary fiber and resistant starch (RS) content are influenced by genes encoding multiple starch synthases and starch branching enzymes. Multiplex editing of SBE genes in wheat and rice significantly increased amylose and RS levels, with stronger effects in higher‐order mutants (Biswas et al., 2023; Li et al., 2021). In barley, targeting four SS and three SBE genes revealed combinatorial effects on RS content and grain traits (Yang et al., 2024). While full knockouts were not recovered, polygenic mutants showed enhanced RS and, in some cases, partial rescue of yield penalties (Yang et al., 2024). This suggests that multiplex editing strategies can be used to create and select lines with improved dietary fiber quality while minimizing negative effects on grain production.
In Nicotiana benthamiana, two β‐1,2‐xylosyltransferase genes and four α‐1,3‐fucosyltransferase genes were simultaneously edited to modify N‐glycosylation patterns in plant‐derived therapeutic proteins (Table 1). The resulting sextuple mutants produced recombinant glycoproteins lacking plant‐specific α‐1,3‐fucose and β‐1,2‐xylose residues, with antibody‐binding behavior comparable to that from mammalian systems (Jansing et al., 2019). The rapid modifications enabled by CRISPR multiplex editing hold promise for overcoming limitations in developing plant‐based glycoproteins for therapeutic applications.
In black poplar (Populus trichocarpa), a predictive modeling approach was used to evaluate over 69,000 possible combinations of manipulations across 21 lignin biosynthetic genes aimed at improving wood properties (Sulis et al., 2023). Seven multiplex editing strategies, each targeting three to six genes, were selected for in planta testing. These strategies were predicted to reduce total lignin content and increase the syringyl‐to‐guaiacyl lignin ratio without negatively affecting plant growth. Although complete knockouts of all target genes were not achieved, some edited lines exhibited improved cellulose‐to‐lignin ratios and enhanced pulping efficiency (Sulis et al., 2023). This work highlights the potential of integrating computational pathway modeling with multiplex editing for complex trait optimization.
In soybean, a CRISPR library targeting 102 genes and their paralogs enabled pooled transformation and trait screening (Bai et al., 2020). This approach uncovered synergistic mutation combinations affecting nodulation without compromising growth (Bai et al., 2020). For species with low transformation rates, this library‐based strategy offers both scalability and a streamlined path for trait discovery, especially when primary transformants can be crossed to increase the mutation diversity. However, its application remains challenging in long‐lived perennials or clonally propagated species due to difficulties in maintaining and propagating mutant lines.
Facile trait stacking and de novo domestication
Multiplex gene editing is particularly well‐suited for stacking desirable alleles spanning multiple traits (Box 1). In rice, thermosensitive male sterility and disease resistance were simultaneously engineered into a breeding line (Li et al., 2019). Transgene‐free, homozygous triple mutants were obtained in the T_1_ generation to accelerate hybrid rice development (Table 1) (Li et al., 2019). Similarly, editing six agronomic genes in an elite wheat variety enabled targeted mutagenesis at up to 15 loci (Luo et al., 2021). This approach allowed rapid pyramiding of favorable alleles controlling multiple traits in hexaploid wheat within a single year, representing a substantial time savings over traditional breeding methods (Luo et al., 2021).
In maize, the BREEDIT pipeline combines multiplex genome editing with conventional crosses to generate over 1000 edited lines targeting 48 growth‐related genes using four 12‐plex constructs (Lorenzo et al., 2023). This approach enabled allele stacking, the recovery of fixed mutations, and the generation of new edits through strategic crosses (Figure 1). Some lines exhibited significant increases in leaf size and drought tolerance (Lorenzo et al., 2023). A similar strategy was applied to a new rice cultivar, FXZ, which has high storage quality and disease resistance but suffers from slow maturation and low adaptability (Wei et al., 2024). Multiplex editing of 12 agronomic genes and rational crosses produced a segregating population with diverse phenotypes. Selected plants exhibited improved early heading and plant architecture without compromising yield or disease resistance (Wei et al., 2024).
Multiplex editing also enables de novo domestication of crop wild relatives by directly targeting domestication genes while preserving beneficial traits to expand gene pools (Kumar et al., 2022; Lemmon et al., 2018; Wolter et al., 2019; Zsögön et al., 2018). Yu et al. (2021) developed a strategic roadmap integrating genome sequencing, efficient transformation, and multiplex editing for de novo domestication of wild allotetraploid rice (Oryza alta) (Figure 1). Using a combination of loss‐of‐function (via single or multiplex mutagenesis) and gain‐of‐function (via base editing) approaches, the team achieved rapid improvements in traits such as seed shattering, grain size, and heading date, while retaining stress resilience from wild germplasm (Yu et al., 2021). This strategy offers a fast‐track route to the development of new crop varieties for enhanced resistance to biotic and abiotic stresses under changing climate conditions.
In long‐lived species like poplar, multiplex editing can circumvent long generation times to accelerate research discovery. An in vitro flowering system was developed by knocking out two paralogs of CEN (Centroradialis), a key floral repressor (Ortega et al., 2023). This fast‐track system is more efficient than heat‐inducible FT (FLOWERING LOCUS T) methods (Hoenicka et al., 2016) and allows flexible allele pyramiding for floral trait investigation. For instance, simultaneous editing of ARR17 (a type‐A response regulator and the main sex regulator) (Müller et al., 2020) and CEN genes induced male flower development in vitro. Allele stacking with edits in trichome‐regulating MYBs yielded glabrous female mutants with hairless seeds (Ortega et al., 2023). These findings revealed shared regulatory pathways between vegetative and reproductive (seed) trichomes and offer promising targets for genetic containment and allergen reduction in urban and plantation forestry.
Eradication of endogenous viruses
Multiplex editing has also opened new avenues for the eradication of endogenous viruses, a breakthrough first demonstrated in porcine cells to inactivate porcine endogenous retroviruses, which are major obstacles in xenotransplantation (Yang et al., 2015). Similar endogenous viral elements in crops, particularly those propagated vegetatively, can severely disrupt breeding programs (Staginnus & Richert‐Pöggeler, 2006). A notable example is the endogenous banana streak virus (eBSV), which has triggered recent outbreaks of banana streak mosaic disease. These outbreaks originated from breeding lines and micropropagated hybrids of Musa accuminata (A genome) and Musa balbisiana (B genome), the latter being a known reservoir of integrated eBSV sequences (Gayral et al., 2008). This genomic integration has significantly limited the use of Musa balbisiana in banana and plantain breeding. Tripathi et al. (2019) used CRISPR multiplex editing to target three ORFs of eBSV in plantain cultivar Gonja Manjaya (AAB genome) and achieved high mutation rates and symptom suppression in several edited lines (Figure 1). DNA analysis indicated that simultaneous disruption of all three ORFs was key to effective virus inactivation (Tripathi et al., 2019). While long‐term monitoring is still needed, this study demonstrates a promising strategy for generating virus‐free lines in primary transformants of vegetatively propagated crops.
BEYOND GENE KNOCKOUTS
Regulatory sequence editing
Multiplex promoter editing, first demonstrated in tomato by Rodríguez‐Leal et al. (2017), is a powerful strategy for fine‐tuning gene expression without prior knowledge of cis regulatory elements. Random mutagenesis, including large deletions, at multiple upstream target sites can produce wide‐ranging effects on gene expression, extending beyond traditional gene knockout or activation. When applied to regulatory genes such as transcription factors (TFs), promoter edits can influence not only the expression of the specific TF but also its upstream regulators, downstream targets, and other interacting partners. These cascading effects may include changes in TF dosage, timing, and spatial patterns of expression and can result in complex phenotypic outcomes that are not necessarily correlated with the expression level of the target gene alone. When combined with selfing or backcrossing, this ‘multiplex mutagenesis drive system’ can rapidly generate large mutant populations with a continuum of genetic variation for quantitative trait engineering (Figure 1) (Hendelman et al., 2021; Liu et al., 2021; Rodríguez‐Leal et al., 2017; Wang et al., 2021). The strategy is also applicable to UTRs and introns that may harbor cis regulatory elements (Table 1) (Song et al., 2022; Zeng et al., 2020).
Multiplex editing of regulatory sequences has yielded novel or beneficial traits such as modified tomato fruit size (Aguirre et al., 2023; Rodríguez‐Leal et al., 2017), multiple ear traits linked to increased maize grain yield (Liu et al., 2021), improved rice grain quality with desirable amylose content (Zeng et al., 2020), as well as increased tiller number and panicle size (Song et al., 2022). Many of these traits have been difficult to stack through conventional breeding due to time constraints, tradeoffs, and linkage drag. Multiplex editing of regulatory elements offers an efficient means to overcome these limitations and disentangle complex trait interactions.
Beyond trait improvement, this approach has advanced our understanding of key developmental circuits. For example, it has been used to dissect the Clavata–Wuschel (CLV‐WUS) pathway regulating stem cell proliferation in tomato (Rodriguez‐Leal et al., 2019; Rodríguez‐Leal et al., 2017; Wang et al., 2021) and maize (Liu et al., 2021), and to explore epistatic relationships among the novel fruit size alleles (Aguirre et al., 2023). An allelic series generated through multiplex editing revealed previously unknown pleiotropic roles of WOX9 (WUS‐related homeobox 9) during vegetative and reproductive development conserved across tomato, groundcherry, and Arabidopsis (Hendelman et al., 2021). Notably, distinct pleiotropic functions were associated with specific cis regulatory regions (Hendelman et al., 2021), underscoring the power of regulatory sequence editing to partition gene function in ways not possible with null mutations.
In rice, editing of IPA1 (Ideal Plant Architecture 1) regulatory elements led to the discovery of a 54 bp promoter deletion that underlies the tradeoff between tiller and panicle number (Song et al., 2022). Mechanistic studies identified a motif within this region as the binding site for An‐1, a basic helix–loop–helix transcription factor that represses IPA1 expression in panicles and roots (Luo et al., 2013; Song et al., 2022). These examples illustrate how multiplex editing of regulatory sequences enables both precision trait engineering and mechanistic dissection of complex gene networks.
Epigenetic and transcriptional regulation
The multiplex CRISPR system has been adapted for transcriptional regulation and epigenetic modification without altering DNA sequences (Box 1). Recent reviews have summarized the development of effector domains and strategies for their recruitment in plant systems (Gardiner et al., 2022; McCarty et al., 2020). The core design uses a catalytically dead Cas9 (dCas9) for site‐specific targeting via multiple gRNAs, with effectors either fused directly to dCas9 or recruited through systems such as SunTag (Tanenbaum et al., 2014), MoonTag (Casas‐Mollano et al., 2023), or modified gRNA scaffolds (Konermann et al., 2015).
Effectors for epigenetic modifications include catalytic domains from DNA methyltransferases (Ghoshal et al., 2021), demethylases (Gallego‐Bartolomé et al., 2018), and histone acetyltransferases of bacterial, mammalian, or plant origin (Lee et al., 2019; Roca Paixão et al., 2019; Selma et al., 2019; Wang, Liu, et al., 2024). These tools modulate promoter accessibility and can fine‐tune gene expression with potentially fewer off‐target effects than direct transcriptional activation (Gardiner et al., 2022). For example, DNA demethylation at the FLOWERING WAGENINGEN (FWA) promoter using a single gRNA significantly upregulated FWA expression and delayed flowering in Arabidopsis (Gallego‐Bartolomé et al., 2018). In contrast, targeted methylation directed by multiple gRNAs led to FWA silencing and early flowering (Ghoshal et al., 2021) (Figure 1). Importantly, the modified DNA methylation states and associated phenotypes were heritable, even in transgene‐free progeny (Gallego‐Bartolomé et al., 2018; Ghoshal et al., 2021). Despite these promising findings, further optimization is needed to enhance efficiency and specificity, particularly with regard to CG, CHG, and CHH contexts, and to ensure long‐term stability of these modifications (Gardiner et al., 2022).
CRISPR‐based transcriptional activation (CRISPRa) and interference (CRISPRi) use effector domains to modulate gene expression without editing DNA. Advanced CRISPRa systems combine dCas9‐activator fusions with MS2 aptamer‐containing gRNA scaffolds and/or SunTag‐based multimeric recruitment (Pan et al., 2021; Selma et al., 2022). Multiplex CRISPRa targeting up to six steps in the flavonoid biosynthetic pathway can selectively induce different flavonoid classes (Selma et al., 2022), including in a cell‐type‐specific manner (Houbaert et al., 2025). A recent study used inducible multiplex CRISPRa for large‐scale screening of morphogenic regulators to enhance plant regeneration in alfalfa (Medicago sativa), strawberry (Fragaria vesca), and sheepgrass (Leymus chinensis) (Zhang et al., 2024). Similarly, a copper‐inducible system enabled tunable activation of metabolic genes in Nicotiana benthamiana (Garcia‐Perez et al., 2022).
MoonTag, with its peptide‐nanobody design, has shown improved performance over SunTag in stable transformants (Casas‐Mollano et al., 2023). Replacing dCas9 with dCas12b or the near‐PAMless dSpRY variant expands the targeting space (Pan et al., 2021). CRISPR‐Combo enables simultaneous editing and activation (Pan et al., 2022) by using Cas9 with long gRNAs (18–20 nt) for cleavage and short gRNAs (≤16 nt) with modified scaffolds for binding without cleavage (Kiani et al., 2015). This allows a single Cas enzyme—whether Cas9, PAMless SpRY, or base editor derivatives—to perform multiplex editing and gene activation across species (Pan et al., 2022). Applications included editing agronomic genes while activating FT for rapid generation cycling in Arabidopsis or morphogenic regulators for accelerated regeneration in poplar and rice (Table 1) (Pan et al., 2022).
Another strategy uses engineered gRNA scaffolds to recruit cytidine and adenosine deaminases via their cognate binding proteins, enabling concurrent C‐to‐T and A‐to‐G base editing of different genes using a single dCas9 (Li et al., 2020). When integrated with CRISPR‐Combo, this design can support higher‐order multiplexing for simultaneous knockout, gain‐of‐function base substitutions, and transcriptional modulation (activation or silencing) at distinct loci.
Recent screening of plant‐ and pathogen‐derived activator domains identified several that outperform VP64 in SunTag‐ and MoonTag‐based multiplex CRISPRa systems in Arabidopsis and Setaria (Zinselmeier et al., 2024). In contrast, CRISPRi remains less explored in plants (Lowder et al., 2015; Piatek et al., 2015), likely due to the availability of RNAi and artificial miRNA tools. Beyond the commonly used SRDX (Superman Repression Domain X) domain, recent studies have shown effective repression using EAR motifs such as DLN114 and its fusions with SRDX (Xu et al., 2023) and ZAT10 (Khan et al., 2025). A novel repressor domain from the cucumber (Cucumis sativus) non‐canonical histone acetyltransferase TENDRIL‐LESS (CsTEN) has been fused with dCas9 to enable programmable transcriptional repression of a wide range of genes in Arabidopsis and, for the first time, in cucumber plants (Figure 1) (Wang, Liu, et al., 2024). Multiplex CRISPRi has also been used to construct reversible gene circuits for spatiotemporal regulation in plants (Khan et al., 2025).
CHROMOSOMAL ENGINEERING
Multiplex CRISPR toolkits have been leveraged to induce targeted chromosomal rearrangements, including inversions, translocations, and arm exchanges (Box 1). In maize, simultaneous induction of double‐strand breaks (DSBs) on opposite sides of the centromere generated a 75.5 Mb pericentric inversion on chromosome 2 (Schwartz et al., 2020). In Arabidopsis, two DSBs near the telomeric ends of chromosome 2 led to a heritable ~17 Mb inversion, retaining only 2 Mb and 0.5 Mb of the original telomeric ends (Rönspies et al., 2022). Multiplex editing also enabled reversal of the 1.1 Mb heterochromatic knob (hk4S) inversion on chromosome 4—an ancient rearrangement estimated to have occurred ~5000 years ago in some Arabidopsis accessions (Schmidt et al., 2020). Crossing the rearranged Col‐0 with Ler‐1 (which lacks the inversion) restored meiotic crossovers in a previously recombination‐suppressed region (Figure 1) (Schmidt et al., 2020). Additional studies, also in Arabidopsis, demonstrated heritable megabase‐scale arm exchanges between chromosomes 1 and 2, and 1 and 5, following multiplex‐induced DSBs (Beying et al., 2020). In both cases, the chromosome‐rearranged Arabidopsis lines were morphologically indistinguishable from the wild type (Beying et al., 2020; Schmidt et al., 2020). However, restoration of meiotic recombination in otherwise suppressed regions represents a notable phenotypic outcome with potential implications for breeding and trait discovery.
A visual assay using a hemizygous betalain pigment marker was developed to help detect rare somatic crossovers in tomato. This system confirmed that multiplex‐induced DSBs can trigger a spectrum of structural changes, including crossovers, chromosomal loss, and chromothripsis‐like rearrangements (Samach et al., 2023). Notably, megabase‐scale inversions had minimal impact on global gene expression and epigenetic state in Arabidopsis (Khosravi et al., 2025). This suggests that targeted inversions could be a powerful tool for fixing beneficial haplotypes and suppressing unwanted recombination in crop breeding.
Multiplex CRISPR has also been adapted for cell‐type‐specific ablation. The CRISPR‐Kill system targets repetitive 45S ribosomal RNA (rDNA) genes to induce cell death (Table 1) (Schindele et al., 2022). When driven by tissue‐specific promoters, CRISPR‐Kill enabled selective elimination of petal cells or reduction of lateral roots in Arabidopsis (Schindele et al., 2022). Coupled with a chemically inducible system, CRISPR‐Kill also allows temporal control of cell ablation (Gehrke et al., 2023), offering new tools for developmental studies and synthetic biology.
TRANSGENE‐FREE EDITED CROPS
From protoplast delivery to agrobacterium transient expression
One of the biggest challenges facing gene‐edited crops is the regulatory framework governing transgenic plants, which hinders not only commercialization but also field trials essential for trait evaluation (Boerjan & Strauss, 2024; Ivanov et al., 2025). This emphasizes the need for transgene‐free CRISPR‐Cas technologies. DNA‐free editing was first demonstrated through direct delivery of preassembled Cas9–gRNA ribonucleoproteins (RNPs) into protoplasts for plant regeneration (Woo et al., 2015). This method was later adapted to generate canker‐resistant citrus (Citrus sinensis) via Cas12a single or multiplex editing of the susceptibility gene LOB1 (lateral organ boundaries) in embryogenic protoplasts (Su et al., 2023; Su et al., 2024). These edited, non‐transgenic citrus plants marked a milestone in agricultural CRISPR applications, receiving United States Department of Agriculture (USDA) Animal and Plant Health Inspection Service (APHIS) and Environmental Protection Agency (EPA) clearance for field release (Su et al., 2023). However, protoplast regeneration remains inefficient in many crop species.
As an alternative, Agrobacterium‐mediated transient expression of multiplex CRISPR constructs, combined with both positive and negative selection, has shown promise across diverse species. Base editing or gene targeting introduces gain‐of‐function mutations in ALS (acetolactate synthase) to confer resistance to sulfonylurea herbicides like chlorsulfuron (Alquézar et al., 2022; Van den Broeck et al., 2025), while simultaneously targeting gene(s) of interest for mutagenesis (Su et al., 2024; Hoengenaert et al., 2025). When paired with a scorable reporter such as GFP, this system enables negative selection against T‐DNA integration events (Figure 1) (Huang et al., 2023). This design has significantly improved the recovery of transgene‐free, biallelic mutants in tobacco, tomato, potato, and citrus in the T_0_ generation by up to two orders of magnitude compared to standard Agrobacterium transient expression without selection (Huang et al., 2023).
In poplar, a cytosine base editor was transiently expressed via Agrobacterium along with multiple gRNAs targeting two endogenous ALS genes and the lignin biosynthesis gene CCoAOMT1 (caffeoyl*‐CoA O‐methyltransferase1*). Nearly half of the chlorsulfuron‐resistant shoots were transgene‐free, although editing efficiency at the lignin gene was low (Hoengenaert et al., 2025). Incorporating a counter‐selection marker could streamline identification of transgene‐free events and reduce downstream screening efforts. Another study reported the efficient co‐editing of ALS and CEN1 genes, resulting in transgene‐free, early‐flowering poplar (Table 1) (Wu et al., 2025). Together, these findings demonstrate that transgene‐free genome editing can be achieved in the first generation using Agrobacterium‐mediated transient expression, offering a scalable and cost‐effective strategy for perennial crops.
Crossing with doubled haploid induction
In annual crops, transgene‐free edited plants are often obtained by segregating out transgenes through crossing, as discussed in earlier sections. However, somatic chimerism remains a challenge when dealing with multiplex or heterozygous edits, often necessitating multiple generations of crossing and screening to obtain homozygous mutants (Box 2). Combining multiplex CRISPR editing with doubled haploid induction offers a promising solution to reduce chimerism and accelerate the development of homozygous mutant lines.
Doubled haploid production, either via in vitro microspore or anther culture, or through haploid inducer lines, can rapidly fix edited alleles by generating homozygous individuals from haploid cells (Qu et al., 2024). When integrated with multiplex editing, this approach enables immediate recovery of uniform, non‐chimeric, and transgene‐free plants, as demonstrated in wheat, maize, and Arabidopsis (Impens et al., 2023; Kelliher et al., 2019; Wang et al., 2019). Haploid inducer lines carrying CRISPR reagents can directly edit elite cultivars that are otherwise recalcitrant to transformation, including intergeneric crosses between wheat ovules and maize inducer pollen, followed by embryo rescue (Kelliher et al., 2019). Alternatively, multiplex‐edited T_0_ plants, such as those derived from the BREEDIT pipeline (Lorenzo et al., 2023), can be backcrossed to wild type to generate heterozygous, segregating populations. Transgene‐free individuals can then be crossed with haploid inducers to recover homozygous mutants with diverse combinations of target gene edits, thereby accelerating quantitative trait improvement (Impens et al., 2023). However, applying this strategy to perennial crops will require a rapid‐cycle flowering system to overcome long generation times.
TECHNICAL CONSIDERATIONS AND CHALLENGES
Multiplex construct design
Our literature mining revealed that the vast majority of multiplex genome editing studies in plants have utilized Cas9 (including nCas9, dCas9, or PAMless variants), with relatively few using Cas12a or Cas12b (Huang et al., 2023; Jordan et al., 2021; Nagy et al., 2025; Pan et al., 2021; Su et al., 2023; Su et al., 2024) (Table 1, Figure 2). This reflects the high efficiency and stability of Cas9 across diverse plant species and applications, from DNA cleavages to transcriptional regulation. However, it also highlights limitations, such as Cas9's large size, which poses challenges for virus‐based vectors with cargo constraints. While the near‐PAMless SpRY variant has expanded the targeting space, slight sequence biases persist (Walton et al., 2020). Exploring the CRISPR‐Cas atlas featuring artificial intelligence (AI)‐generated editors (Ruffolo et al., 2025) can further expand the diversity of Cas variants for plant applications (Box 2). Continued development of multiplex editing toolkits should explore smaller Cas or Cas‐like proteins to broaden delivery options. Incorporating tissue‐specific or inducible promoters (Bollier et al., 2021; Decaestecker et al., 2019; Garcia‐Perez et al., 2022; Gehrke et al., 2023; Zhang et al., 2024) will also enable spatiotemporal control of Cas activity.
Multiplex CRISPR genome editing reagents and mutation mapping.The left panel depicts commonly used Cas proteins, including Cas9, Cas12a, and Cas12b. The middle panel illustrates various strategies for gRNA expression using different polymerase III (Pol III), Pol II, and hybrid configurations. gRNA can be processed by tRNA‐, ribozyme‐ or Csy4‐based systems. The right panel shows post‐editing mutation detection approaches, including PCR followed by Sanger sequencing, amplicon sequencing via two‐step PCR with short‐read sequencing, and long‐read sequencing for broader genomic analysis.
As multiplex editing increases in complexity, so does the number of gRNAs that must be expressed. For CRISPR‐Cas9 systems, two common approaches are used: (1) each gRNA is driven by its own promoter, typically a Pol III (Polymerase III) promoter, or (2) a single (Pol II or Pol III) promoter drives expression of a polycistronic gRNA array, with individual gRNAs separated by cleavable RNA elements such as tRNAs (Xie et al., 2015), self‐cleaving ribozymes (Nowak et al., 2016), or recognition sequences for exogenous nucleases like Csy4 (Qi et al., 2012) (Table 1, Figure 2). The first approach allows independent control of each gRNA, and constructs are often built using Gateway or Golden Gate cloning systems and their derivatives (Čermák et al., 2017; Lowder et al., 2015; Ma, Zhang, et al., 2015; Marillonnet & Grützner, 2020; Sarrion‐Perdigones et al., 2011; Zhang et al., 2020). However, repeated use of the same Pol III promoter or inclusion of long promoter sequences can make constructs bulky and difficult to assemble. Recent studies have shown that minimal Pol III promoters—70 bp for dicots (Deguchi et al., 2025) and 160 bp for monocots (Nagy et al., 2025)—are effective for gRNA expression. Using short, non‐redundant promoters not only reduces construct size but also streamlines cloning by enabling low‐cost DNA synthesis for one‐step assembly of multiplex gRNA cassettes directly into binary vectors (Deguchi et al., 2025; Nagy et al., 2025; Ortega et al., 2023).
The second approach uses a single promoter to express multiple gRNAs, simplifying transcriptional control. However, this design inevitably introduces other repetitive elements, such as tRNAs, ribozymes, or Csy4, for gRNA processing, which can offset gains in compactness. The efficiency of these gRNA processing systems can vary by species and construct architecture, requiring empirical optimization (Čermák et al., 2017; Cao et al., 2022). Furthermore, not all gRNAs are equally effective (Table 1) (de Vries et al., 2021; Houbaert et al., 2025; Ma, Yang, et al., 2024; Sánchez‐León et al., 2024; Yang et al., 2024). In a few studies, editing or activation efficiencies declined as the number of gRNAs increased (Angulo et al., 2023; Selma et al., 2022; Stuttmann et al., 2021; Xie et al., 2015), though the underlying causes remain unclear. A drop in multiplex editing efficiency was also reported in soybean transformed via a pooled library with a simple and identical construct architecture (one gRNA per construct) (Bai et al., 2020), although potential silencing could not be ruled out in this case due to integration of multiple T‐DNA copies. Unlike Cas9, Cas12a (Cpf1) can self‐process pre‐crRNA arrays, eliminating the need for additional processing elements (Jordan et al., 2021). Nonetheless, independent promoters, tRNA‐gRNA, and ribozyme‐based methods have also been used with Cas12a for multiplex editing (Huang et al., 2023; Nagy et al., 2025).
A less explored yet essential component of CRISPR‐Cas9 multiplex constructs is the gRNA scaffold—a synthetic fusion of crRNA and tracrRNA required for Cas9 binding (Nishimasu et al., 2014). Because each gRNA includes a scaffold, this element becomes highly repetitive in multiplex designs. In E. coli, Reis et al. (2019) developed a high‐throughput design‐build‐test‐learn framework that integrated biophysical modeling, experimental characterization, and machine learning (ML) to systematically optimize gRNA scaffold variants using 23 design principles. This approach enabled DNA synthesis and seamless assembly of CRISPR arrays containing up to 22 non‐redundant gRNAs, overcoming scaffold repetitiveness as a key bottleneck in multiplex editing. Similar strategies have been applied in plants, where structure‐guided and random mutagenesis approaches have yielded diverse, functional scaffold variants (Wang, Li, et al., 2024).
The growing adoption of AI and large language models (LLMs) in biological research will further advance CRISPR innovation. For example, the recently developed CRISPR‐GPT enables LLM‐powered automation of the entire CRISPR experimental workflow, from design and planning to execution and data analysis (Qu et al., 2025). Its modular, multi‐agent architecture facilitates human‐AI collaboration, streamlining complex multiplex genome engineering, as demonstrated in proof‐of‐concept gene knockout and epigenetic activation experiments in human cell lines (Qu et al., 2025). Expanding CRISPR‐GPT applications to agricultural crops holds tremendous promise for broadening access to precision genome editing across diverse plant systems (Box 2).
Mutation determination
A decade ago, when CRISPR was first applied to plants, mutation detection often relied on targeting regions with restriction enzyme sites or designing two gRNAs per gene. This enabled simple PCR‐based genotyping, sometimes followed by restriction enzyme or T7 endonuclease I digestion, before confirming edits via Sanger sequencing (Nekrasov et al., 2013; Shan et al., 2013; Wang, Naik, et al., 2014) (Table 1, Figure 2). Direct sequencing of PCR products was also common, and several computational tools were developed to interpret mixed chromatograms (Brinkman et al., 2014; Conant et al., 2022; Ma, Chen, et al., 2015). While rapid and cost‐effective for small‐scale validation, PCR‐based methods lack the resolution to detect subtle edits or distinguish among multiple gene copies or alleles. Multi‐tier PCR genotyping has been used to identify inversion alleles from TAG editing (Liu et al., 2023), but this modified approach is labor‐ and time‐intensive, requiring context‐dependent primer design that is difficult to scale and ineffective for other structural rearrangements. Sanger sequencing is also inadequate for detecting low‐frequency or complex edits, especially in chimeric tissues, polyploid genomes, or multiplex experiments. Cloning PCR products prior to sequencing can improve allelic resolution but is more resource‐intensive and not scalable.
Amplicon sequencing, typically with the Illumina platform, offers high‐throughput, scalable detection of mutation patterns and editing efficiency at individual target sites (Table 1, Figure 2) (Li et al., 2018; Sánchez‐León et al., 2018; Schmidt et al., 2019; Tang et al., 2017; Wang et al., 2018; Woo et al., 2015; Zhou et al., 2015). For multi‐allelic targets, large deletions that disrupt primer binding sites can be inferred as no‐amplification alleles (Bewg et al., 2022; Chen et al., 2023). When consensus primers are used to amplify highly homologous regions, both on‐ and off‐target effects can be assessed simultaneously (Tsai et al., 2020; Zhou et al., 2015). Several open‐source tools are available to facilitate high‐throughput data processing (Bruyneel et al., 2019; Li et al., 2025; Liu et al., 2019; Park et al., 2016; Xue & Tsai, 2015). A recent review provides additional computational resources, including gRNA design tools, for large‐scale CRISPR experiments (Huang et al., 2022). However, the short‐read length of the Illumina platform (e.g., 2 × 150 bp or 2 × 250 bp) is a limitation, especially when distinguishing between highly homologous paralogs and alleles that lack sufficient sequence variation, such as single‐nucleotide polymorphisms (SNPs). This constraint can limit the available sequence space for gRNA design. Short‐read sequencing is also unable to detect large or complex structural changes, such as inversions and translocations, which are often associated with TAG editing, as discussed earlier.
Target capture sequencing offers a PCR‐free alternative that enables de novo assembly and detection of unexpected structural variants, including complex inversions (Table 1) (Chen et al., 2023), within the genomic regions of interest. However, it is more resource‐intensive and may not be practical for routine screening of large populations. Whole‐genome short‐read sequencing has been used to detect chromosomal translocations in Arabidopsis, which has a small genome and high‐quality reference (Table 1) (Beying et al., 2020). This approach is less feasible for heterozygous or polyploid species.
Although not yet widely adopted for CRISPR mutational mapping (Sato et al., 2024), long‐read sequencing technologies—especially the portable and cost‐effective MinION and Flongle devices from Oxford Nanopore Technologies (ONT)—are emerging as powerful tools for structural variation detection in multiplex experiments (Figure 2). When combined with adaptive sampling, these platforms can enable real‐time, targeted enrichment of regions of interest (Martin et al., 2022). With improvements in error rate and sufficient read coverage, ONT sequencing can also detect SNPs and small indels (Sato et al., 2024), making it a promising platform for comprehensive analysis of multiplex editing outcomes.
Whole‐genome sequencing provides the most comprehensive view of both on‐ and off‐target effects. However, it remains cost‐prohibitive for most plant species, particularly those lacking high‐quality reference genomes. The common practice has been to sequence one or a few edited events (Table 1) (Beying et al., 2020; Hendelman et al., 2021; Huang et al., 2023; Jordan et al., 2021; Li et al., 2020; Peterson et al., 2016; Rodríguez‐Leal et al., 2017; Su et al., 2024; Van den Broeck et al., 2025; Wei et al., 2024; Wu et al., 2025; Yu et al., 2024). Even then, off‐target analysis is typically limited to computationally predicted sites, as genome‐wide assessments are confounded by spontaneous mutations and natural polymorphisms. When off‐target potential is minimized during gRNA design, whole‐genome sequencing of edited plants may not be necessary. However, for complex multiplexing or chromosomal engineering, whole‐genome sequencing or optical genome mapping (Table 1) is essential to validate editing outcomes (Schwartz et al., 2020).
Finally, our review of recent publications highlights a shift in the CRISPR field—from system optimization and method validation to its routine use in biological research as a standard and reliable tool. Mutation mapping details and editing metrics are no longer the focal point, but are often abbreviated as part of the routine process of generating a few confirmed mutants to support downstream functional characterization (Table 1) (Shen et al., 2019; Shen et al., 2022; Yu et al., 2021; Zhong et al., 2019). In this evolving context, multiplex editing studies increasingly rely on trait‐based screening rather than exhaustive genotyping, especially when editing outcomes are complex or when phenotypic consequences cannot be reliably inferred from mutation patterns alone. This is particularly central for regulatory sequence editing and multiplex experiments, where combinatorial effects and intermutant interactions may yield unexpected or emergent traits (Lorenzo et al., 2023; Song et al., 2022; Wei et al., 2024; Yang et al., 2024).
CONCLUSIONS AND FUTURE OUTLOOK
Multiplex gene editing is reshaping the landscape of plant biotechnology and crop improvement. It offers an efficient and scalable alternative to traditional breeding and single‐gene editing by enabling simultaneous and combinatorial manipulation of complex traits. As toolkits become more sophisticated and accessible, and as analytical pipelines improve for detecting complex editing outcomes, multiplex editing is poised to become a core technology in both basic research and applied breeding programs.
Beyond editing genes and regulatory sequences, multiplex approaches will be instrumental in targeting highly repetitive and previously intractable genomic regions, such as transportable elements (Guo et al., 2024), satellite or tandem repeats (Schindele et al., 2022; Tripathi et al., 2019), and heterochromatin (Khosravi et al., 2025; Weiss et al., 2022). These regions hold untapped potential for understanding genome function and unlocking new avenues for trait innovation.
Future efforts should focus on improving gRNA efficiency prediction to enhance editing performance, minimizing off‐target effects, and developing robust and species‐agnostic delivery systems, especially for recalcitrant, perennial, and/or polyploid species. The growing integration of AI, ML, and LLMs into plant breeding and bioengineering research (Farooq et al., 2024; Lam et al., 2024; Yan & Wang, 2022; Zhang et al., 2025) offers powerful tools to streamline experimental design, optimize construct assembly strategy, and predict editing outcomes. These technologies can accelerate every stage of the synthetic biology and plant design workflow, from target gene and gRNA selection to genetic part compatibility, construct building, assay design, and phenotyping, thereby enhancing the speed and precision of design‐build‐test‐learn cycles (Qu et al., 2025). With continued innovation in CRISPR reagents, synthetic biology toolkits, delivery technologies, and computational design pipelines, multiplex editing holds immense promise for harnessing the full potential of plant genome engineering to meet the challenges of sustainable agriculture, food security, energy independence, and climate resilience.
Conflict of Interest
The authors declare no conflict of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Aguirre, L. , Hendelman, A. , Hutton, S.F. , Mc Candlish, D.M. & Lippman, Z.B. (2023) Idiosyncratic and dose‐dependent epistasis drives variation in tomato fruit size. Science, 382, 315–320.37856609 10.1126/science.adi 5222 PMC 10602613 · doi ↗ · pubmed ↗
- 2Alquézar, B. , Bennici, S. , Carmona, L. , Gentile, A. & Peña, L. (2022) Generation of transfer‐DNA‐free base‐edited citrus plants. Frontiers in Plant Science, 13, 835282.35371165 10.3389/fpls.2022.835282 PMC 8965368 · doi ↗ · pubmed ↗
- 3Angulo, J. , Astin, C.P. , Bauer, O. , Blash, K.J. , Bowen, N.M. , Chukwudinma, N.J. et al. (2023) CRISPR/Cas 9 mutagenesis of the Arabidopsis GROWTH‐REGULATING FACTOR (GRF) gene family. Frontiers in Genome Editing, 5, 1251557.37908969 10.3389/fgeed.2023.1251557 PMC 10613670 · doi ↗ · pubmed ↗
- 4Assaad, F.F. , Tucker, K.L. & Signer, E.R. (1993) Epigenetic repeat‐induced gene silencing (RIGS) in Arabidopsis. Plant Molecular Biology, 22, 1067–1085.8400126 10.1007/BF 00028978 · doi ↗ · pubmed ↗
- 5Bai, M. , Yuan, J. , Kuang, H. , Gong, P. , Li, S. , Zhang, Z. et al. (2020) Generation of a multiplex mutagenesis population via pooled CRISPR‐Cas 9 in soya bean. Plant Biotechnology Journal, 18, 721–731.31452351 10.1111/pbi.13239 PMC 7004907 · doi ↗ · pubmed ↗
- 6Bewg, W.P. , Harding, S.A. , Engle, N.L. , Vaidya, B.N. , Zhou, R. , Reeves, J. et al. (2022) Multiplex knockout of trichome‐regulating MYB duplicates in hybrid poplar using a single g RNA. Plant Physiology, 189, 516–526.35298644 10.1093/plphys/kiac 128PMC 9157173 · doi ↗ · pubmed ↗
- 7Beying, N. , Schmidt, C. , Pacher, M. , Houben, A. & Puchta, H. (2020) CRISPR–Cas 9‐mediated induction of heritable chromosomal translocations in Arabidopsis . Nature Plants, 6, 638–645.32451449 10.1038/s 41477-020-0663-x · doi ↗ · pubmed ↗
- 8Biswas, S. , Ibarra, O. , Shaphek, M. , Molina‐Risco, M. , Faion‐Molina, M. , Bellinatti‐Della Gracia, M. et al. (2023) Increasing the level of resistant starch in ‘presidio’ rice through multiplex CRISPR‐Cas 9 gene editing of starch branching enzyme genes. Plant Genome, 16, e 20225.35713092 10.1002/tpg 2.20225 PMC 12806894 · doi ↗ · pubmed ↗