Novel MAML2 Fusions in Human Malignancy
Takefumi Komiya, Kieran Sweeney, Chao H. Huang, Anthony Crymes, Emmanuel S. Antonarakis, Andrew Elliott, Matthew J. Oberley, Mark G. Evans

TL;DR
This study identifies new gene fusions involving MAML2 in human cancers, most likely linked to genomic instability, with one fusion requiring further investigation.
Contribution
The study discovers novel MAML2 fusion partners and characterizes their genomic and mutational contexts.
Findings
Novel MAML2 fusions were found in 143 tumor samples, with most associated with genomic instability and TP53 co-mutations.
Most novel fusions occurred near MAML2 and likely resulted from duplications or deletions, except ATXN3::MAML2 which arose via interchromosomal translocation.
ATXN3::MAML2 fusions were not associated with TP53 mutations and may represent a pathogenic alteration requiring further study.
Abstract
Many gene fusions have been implicated in the development and progression of human cancers. Our group and others have identified specific gene fusions involving the MAML2 gene in human cancers, and we were interested in searching for additional novel MAML2 fusions. Our analysis with the Caris Life Sciences molecular database demonstrated that most of the novel fusions were associated with increased fusion burden and genomic loss of heterozygosity, which are potentially suggestive of genomic instability, except for ATXN3::MAML2 which warrants further investigation. Background: Oncogenic fusions of MAML2 with CRTC1, CRTC3, YAP1, and NR1D1 retain the MAML2 transactivating domain (TAD) and are believed to drive aberrant gene transcription. While the oncogenic roles of these known fusions have been established, we aimed to identify novel MAML2 fusions across a range of human malignancies.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
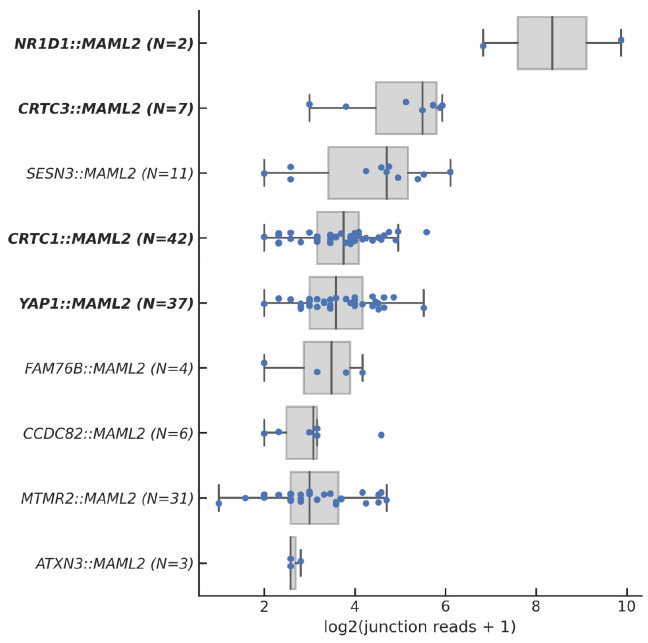 Figure 1
Figure 1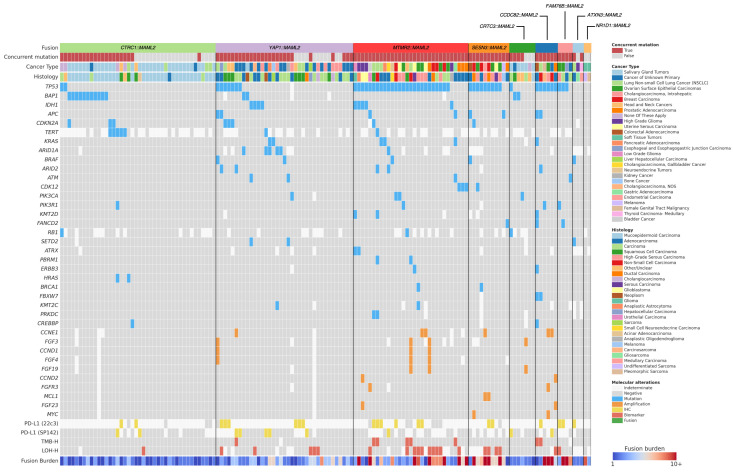 Figure 2
Figure 2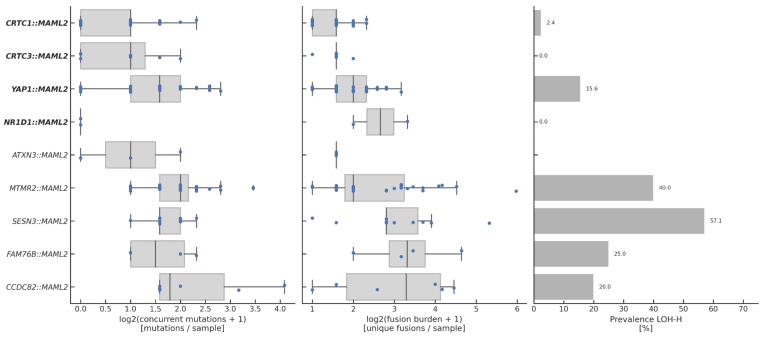 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHippo pathway signaling and YAP/TAZ · Cancer Genomics and Diagnostics · Cancer-related gene regulation
1. Introduction
MAML2 (Mastermind-like 2) is a family member of Mastermind genes that are primarily involved in regulation of the NOTCH signaling pathway. MAML2 activates downstream signaling by binding intra-cellular NOTCH and consists of five exons in which the NOTCH binding domain and transcriptional activation domain (TAD) are located at the N-terminal and C-terminal portions, respectively [1].
Our group was involved in the discovery of a CRTC1::MAML2 fusion gene, which resulted from a t (11; 19) (q14–21; p12–13) translocation in mucoepidermoid carcinoma (MECT) [2]. It typically develops in the parotid gland but can occur in other organs, such as the lungs [3]. The fusion is detected in more than 50% of MECT cases, regardless of primary location, and inhibition of the CRTC1::MAML2 fusion or its downstream targets suppresses cell growth in both in vitro and in vivo models [4,5,6], supporting the notion that CRTC1::MAML2 is a specific driver oncogene in MECT. Further studies also found CRTC3::MAML2 fusions in MECT. Both fusions retain the N-terminal CREB binding domain of CRTC1/3 and the C-terminal TAD of MAML2 and drive oncogenesis via aberrant expression of CREB target genes [7,8,9].
Studies including the one by Valouev A et al. reported frequent in-frame YAP1::MAML2 fusions in nasopharyngeal carcinoma, metaplastic thymoma, poroma, malignant undifferentiated sarcoma, ependymoma, and others [10,11,12,13,14]. YAP1 is a transcriptional coactivator involved in the regulation of the Hippo and Wnt pathways [15]. YAP1::MAML2 fusions retain the N-terminal TEAD-binding region of YAP1 and the C-terminal TAD of MAML2 and have been shown to interact with TEAD transcription factors to activate the YAP1 transcriptional program [16].
In addition to CRTC1/3 and YAP1, other MAML2 fusion partners such as CXCR4 in chronic lymphocytic leukemia, KMT2A in thymoma, and C11orf95 and ZFTA in ependymoma have been reported [17,18,19,20,21]. The NR1D1 partner has also recently been described as an emerging epithelioid and spindle cell sarcoma subtype [22]. All reported fusions retain the MAML2 TAD and do not activate NOTCH signaling due to loss of the C-terminal NOTCH binding site, suggesting that MAML2 fusions activate the downstream signaling of the partner gene rather than the NOTCH pathway. Our study aimed to identify additional previously undescribed fusion partners of MAML2 across a range of human malignancies, detected by whole transcriptome sequencing, in which we identified new oncogenic alterations that could be amenable to targeted therapy in cancer subtypes.
2. Materials and Methods
2.1. Compliance Statement
This retrospective study was reviewed by the institutional review board at Penn State Health and was designated exempt from human subject research. The study was also conducted under Caris Life Sciences’ Research Data Banking protocol, which was reviewed and granted IRB exemption by the WCG IRB. The study adhered to the ethical guidelines of the Declaration of Helsinki, the Belmont Report, and the U.S. Common Rule.
2.2. DNA Sequencing
Genomic DNA was isolated from formalin-fixed paraffin-embedded (FFPE) tumor samples and sequenced using the NextSeq or NovaSeq 6000 platforms (Illumina, Inc., San Diego, CA USA). Tumor enrichment was achieved prior to sequencing by harvesting targeted tissues using manual microdissection techniques. A custom SureSelect XT assay (Agilent Technologies, Santa Clara, CA USA) was used to enrich 592 whole-gene targets for the NextSeq sequenced tumors. Over 700 clinically relevant genes at high coverage and read depth were used for NovaSeq sequenced tumors, along with another panel designed to enrich another 20,000+ genes at lower depth. Variants were detected with an average sequencing depth of coverage of >500, an analytic sensitivity of 5%, and >99% confidence based on allele frequency and amplicon coverage. Identified genetic variants were interpreted by board-certified molecular geneticists and categorized according to the American College of Medical Genetics and Genomics (ACMG) standards. Only pathogenic and likely pathogenic mutations were counted when determining the mutation frequencies of individual genes. The copy number alteration (CNA) of each exon was determined by calculating the average depth of the sample along with the sequencing depth of each exon and comparing the calculated result to a pre-calibrated value.
2.3. Whole Transcriptome Sequencing
mRNA was sequenced using the Illumina NovaSeq platform (Illumina, Inc., San Diego, CA, USA) and Agilent SureSelect Human All Exon V7 bait panel (Agilent Technologies, Santa Clara, CA, USA). RNA was isolated from formalin-fixed paraffin-embedded (FFPE) tumor samples that underwent pathology review to assess tumor content and size; at least 10% of tumor content in the area for microdissection was required for enrichment and extraction of tumor-specific RNA. RNA extraction was performed using a Qiagen RNA FFPE tissue extraction kit. An Agilent TapeStation was used to determine RNA quantity and quality. Synthesized and purified cDNA targets were hybridized to biotinylated RNA baits; a post capture PCR reaction was used to amplify the bait–target complexes. Resultant libraries were quantified and normalized, and the pooled libraries were denatured, diluted and sequenced. GRCh37/hg19 was the reference genome. The test had ≥97% Positive Percent Agreement (PPA), ≥99% Negative Percent Agreement (NPA) and ≥99% Overall Percent Agreement (OPA) with a validated comparator method. Gene fusions were identified from WTS data using STAR-Fusion.
2.4. Loss of Heterozygosity
Genomic loss of heterozygosity (LOH) was determined by splitting the 22 autosomal chromosomes into 552 segments (2–6 Mb in size) and calculating the LOH of single nucleotide polymorphisms (SNPs) within each segment. Student’s t-test was used to compare SNP distances of each region to control distances of a non-LOH reference (NA12878; RRID:CVCL_7526). A region was called positive if the average distance was larger than 0.15 Mb and the corrected p-value was less than 0.02. Segments spanning ≥ 90% of a whole chromosome or chromosome arm and segments not covered by the SNP backbone and the WES panel were excluded from the calculation. Samples were called genomic LOH High (LOH-H) if ≥16% of all 552 segments had LOH or Low if <16% of all 552 segments had LOH.
2.5. Statistical Analysis
Statistical analysis was performed in Python (v.3.10.4) using the Numpy (v.1.23.3), Pandas (v.1.5.1), and Scipy (v.1.12.0) packages. Significance was calculated using Chi-Square, Fisher’s Exact, or Mann–Whitney-U tests.
3. Results
3.1. Overview of Fusions
Among 180,124 pan-cancer tumor samples with DNA and RNA sequencing results, 509 samples (0.3%) harbored a MAML2 fusion. A total of 169 fusions remained after filtering the fusions to retain only those that preserved the C-terminal TAD of MAML2 (Table S1). After further filtering to focus only on recurrent (occurring in ≥3 patients) or known pathogenic or likely pathogenic (P/LP) fusions, a total of 143 fusion-positive tumor samples remained (Table 1). The fusions included known P/LP fusions of MAML2 with CRTC1/3, YAP1, and NR1D1 and unclassified fusions involving MTMR2, SESN3, CCDC82, FAM76B, and ATXN3.
CTRC1::MAML2 and YAP1::MAML2 were the most common P/LP fusions, and MTMR2::MAML2 was the most common unclassified fusion. Excluding YAP1::MAML2, which arose via inversion, the known P/LP fusions featured partner genes on chromosomes other than MAML2 that arose via translocation events. In contrast, the unclassified fusions featured partner genes located near MAML2 on 11q21 and arose from duplications or deletions, except for ATXN3::MAML2, which arose from a t (14; 11) translocation.
Functionally, the P/LP fusion partners CRTC1, CRTC3, YAP1, and NR1D1 were transcriptional activators or repressors. The unclassified fusion partners had diverse functions and no clear role in transcriptional regulation, except for ATXN3, a deubiquitinating enzyme reported to control transcription by regulating chromatin structure [23]. The ATXN3::MAML2 fusions retained most functional domains of ATXN3, with C-terminal breakpoints of its PolyQ region.
3.2. Clinical Characteristics of Fusion-Positive Patients
Patients with recurrent or P/LP MAML2 fusions had a median age of 62 years (range: 17–90+) and were 58% female (Table S2). The CRTC1::MAML2 and CRTC3::MAML2 fusions occurred primarily in mucoepidermoid carcinomas of the salivary gland or the head and neck; both NR1D1::MAML2 fusions were detected in sarcomas. YAP1::MAML2 was appreciated in a relatively broad distribution of cancers; however, a preponderance of these fusion-positive cases arose in the biliary tract (48.7%). Overall, the unclassified MAML2 fusions had no clear association with the tissue of origin (Table 2).
3.3. Expression of MAML2 Fusions
We next assessed the expression levels of the MAML2 fusions (Figure 1). NR1D1::MAML2, CTRT3::MAML2, and SESN3::MAML2 were the most highly expressed fusions, while ATXN3::MAML2 had the lowest expression. Overall, the unclassified fusions had lower expressions than the known P/LP fusions. (8 vs. 13 median junction reads, p = 0.0064, Figure S1).
3.4. Genomic Landscape of Fusion-Positive Tumors
The genomic landscape of fusion-positive tumors varied by fusion isoform is shown in Figure 2. Overall, the most common co-alterations were P/LP mutations of TP53 (N = 54), BAP1 (N = 15), and IDH1 (N = 9). Samples harboring unclassified fusions were enriched in P/LP TP53 mutations compared to those with known P/LP fusions (80.0% vs. 11.5%, p < 0.0001), though, notably, the ATXN3::MAML2 samples had none. More generally, samples harboring unclassified fusions tended to have the most concurrent P/LP mutations (Figure 3, left). In fact, with the notable exception of ATXN3::MAML2, all samples with an unclassified fusion harbored at least one concurrent P/LP mutation, whereas at least 30% of the samples associated with each known P/LP fusion harbored none (Table S3).
To quantify genomic instability, we also assessed fusion burden and genomic loss of heterozygosity (LOH) (Figure 3, middle and right). Fusion burden was quantified as the number of unique fusion isoforms detected per sample and was positively correlated with LOH (Spearman Rho = 0.60, p < 0.0001, Figure S2A). Fusion burden was the highest for fusions involving FAM76B, SESN3, and CCDC82 and the lowest for CRTC1/3 and ATXN3. The prevalence of genomic LOH (LOH-H status) was the highest for fusions involving SESN3, MTMR2, FAM76B, and CCDC82, but was also notably high among YAP1::MAML2 samples. Overall, fusion burden was higher in samples harboring unclassified fusions vs. known P/LP fusions (median: 6 vs. 2 fusion isoforms, p < 0.0001, Figure S2B), as was the prevalence of LOH-H status (39.0 vs. 7.4%, p < 0.0001, Figure S2C).
4. Discussion
We retrospectively analyzed a total of 180,124 tumor samples that underwent comprehensive genetic analysis and found 509 specimens with MAML2 fusions. After filtering out non-recurrent fusions and fusions lacking a MAML2 TAD, 143 specimens remained.
CRTC1/3 and YAP1 were the most common fusion partners of MAML2. While previously reported, tumors demonstrating these translocation events may be amenable to targeted therapy. Recent studies indicate that the CRTC1/3::MAML2 fusions prevalent in mucoepidermoid carcinoma [2,3,8,9] may be susceptible to the effects of CDK4/6 and EGFR inhibitors [6]. Moreover, several pharmacologic agents are currently undergoing early-phase clinical trials targeting solid tumors with disrupted Hippo pathway signaling [24,25], such as NF2-deleted mesothelioma and tumors harboring YAP1 fusions [26,27]. These potential therapies could possibly be employed for the YAP1::MAML2 fusion tumors identified in our study, including those arising in the biliary tract.
Given what is known about the CRTC1/3 and YAP1 fusion partners with MAML2, our study seeks to determine whether additional, previously unreported partner genes exist and have therapeutic or diagnostic implications. Most of the novel, unclassified fusions have low junction reads, high fusion burden, and frequent co-alterations, notably TP53 mutations. Most of the partner genes were located in the region of the MAML2 gene on chromosome 11q22. These fusions were seen in tumors arising from a wide variety of organs and did not cluster in any specific disease. These findings suggest that they are potential passenger alterations—products of chromosomal instability, possibly due to impaired p53 function—and do not seem to have any biologic role in tumorigenesis.
Notably, we found three cases featuring in-frame ATXN3::MAML2 fusions. ATXN3 is involved in the degradation of misfolding chaperone substrates, transcriptional regulation, and maintenance of protein homeostasis [28]. Depletion of ATXN3 leads to spinocerebellar ataxia type 3, an age-related neurodegenerative disease. In preclinical models, deletion of the ATXN3 gene in cancer cells resulted in decreased YAP1 protein levels and decreased expression of YAP1 target genes [29], which raise the possibility of targeting the YAP1/TEAD transcriptional program as a potential therapeutic strategy for these tumors [27]. Moreover, Lee JW et al. has reported a single case of ATXN3::MAML2 fusion in a pancreatic intraductal tubulopapillary neoplasm [30]; this novel finding in a pre-cancerous condition could indicate oncogenicity. In particular, our study showed that ATXN3::MAML2-positive cases exhibited low fusion burdens and were not always associated with alternate driver mutations, including those involving TP53. Ultimately, this novel fusion may warrant further investigation as it occurs in and could be a potential target for human cancers.
Translational/Clinical Relevance
All previously reported pathogenic fusions involving MAML2 contain the C-terminus transactivation domain and lose the N-terminus NOTCH binding site. The molecular outcome of these fusions includes activation of downstream pathways/genes of the MAML2 fusion partner (e.g., CRTC1/3, YAP1). Selective targeting of MAML2 fusions requires inhibition of the interaction between the partner gene and its transcriptional targets, emphasizing the importance of individualized therapeutic intervention. Compared to the previously known MAML2 fusions, our study suggests that rearrangement events with novel gene partners may primarily serve as passenger co-alterations to additional driver mutations, with the exception of ATXN3::MAML2, which may be implicated in oncogenesis with further study.
5. Conclusions
As an expansion to earlier work [31], we identified novel MAML2 fusion partners, most of which had no clear role in transcriptional regulation or association with tissue of origin but were associated with increased fusion burden, increased genomic loss of heterozygosity, and impaired p53 function. Further study is needed to determine if the novel fusions are pathogenic alterations or simply passenger alterations associated with genomic instability. In particular, ATXN3::MAML2 fusions, previously reported in a pre-cancerous pancreatic disease case, may represent a pathogenic alteration warranting further investigation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wu L. Sun T. Kobayashi K. Gao P. Griffin J.D. Identification of a Family of Mastermind-Like Transcriptional Coactivators for Mammalian Notch Receptors Mol. Cell. Biol.2002227688770010.1128/MCB.22.21.7688-7700.200212370315 PMC 135662 · doi ↗ · pubmed ↗
- 2Tonon G. Modi S. Wu L. Kubo A. Coxon A.B. Komiya T. O’Neil K. Stover K. El-Nagger A. Griffin J.D. t(11;19)(q 21;p 13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway Nat. Genet.20033320821310.1038/ng 108312539049 · doi ↗ · pubmed ↗
- 3Saade R.E. Bell D. Garcia J. Roberts D. Weber R. Role of CRTC 1/MAML 2 Translocation in the Prognosis and Clinical Outcomes of Mucoepidermoid Carcinoma JAMA Otolaryngol.–Head. Neck Surg.201614223424010.1001/jamaoto.2015.327026796488 · doi ↗ · pubmed ↗
- 4Komiya T. Park Y. Modi S. Coxon A.B. Oh H. Kaye F.J. Sustained expression of Mect 1-Maml 2 is essential for tumor cell growth in salivary gland cancers carrying the t(11;19) translocation Oncogene 2006256128613210.1038/sj.onc.120962716652146 · doi ↗ · pubmed ↗
- 5Chen Z. Lin S. Li J.-L. Ni W. Guo R. Lu J. Kaye F.J. Wu L. CRTC 1-MAML 2 fusion-induced lnc RNA LINC 00473 expression maintains the growth and survival of human mucoepidermoid carcinoma cells Oncogene 2018371885189510.1038/s 41388-017-0104-029353885 PMC 5889358 · doi ↗ · pubmed ↗
- 6Chen Z. Ni W. Li J.-L. Lin S. Zhou X. Sun Y. Li J.W. Leon M.E. Hurtado M.D. Zolotukhin S. The CRTC 1-MAML 2 fusion is the major oncogenic driver in mucoepidermoid carcinoma JCI Insight 20216 e 13949710.1172/jci.insight.13949733830080 PMC 8119194 · doi ↗ · pubmed ↗
- 7Coxon A. Rozenblum E. Park Y.-S. Joshi N. Tsurutani J. Dennis P.A. Kirsch I.R. Kaye F.J. Mect 1-Maml 2 Fusion Oncogene Linked to the Aberrant Activation of Cyclic AMP/CREB Regulated Genes Cancer Res.2005657137714410.1158/0008-5472.CAN-05-112516103063 · doi ↗ · pubmed ↗
- 8Birkeland A.C. Foltin S.K. Michmerhuizen N.L. Hoesli R.C. Rosko A.J. Byrd S. Yanik M. Nor J.E. Bradford C.R. Prince M.E. Correlation of Crtc 1/3-Maml 2 fusion status, grade and survival in mucoepidermoid carcinoma Oral. Oncol.2017685810.1016/j.oraloncology.2017.02.02528438292 PMC 5433350 · doi ↗ · pubmed ↗