Integrating Molecular Phenotyping into Treatment Algorithms for Advanced Oestrogen Receptor-Positive Breast Cancer
Sarah Childs, Ryoko Semba, Lucy Haggstrom, Elgene Lim

TL;DR
This paper reviews how molecular profiling can improve treatment decisions for advanced estrogen receptor-positive breast cancer by integrating genetic insights into clinical strategies.
Contribution
The paper uniquely integrates molecular phenotyping with clinical treatment algorithms for advanced ER-positive breast cancer.
Findings
Molecular profiling helps identify genomic alterations driving resistance and progression in ER-positive breast cancer.
Oral SERDs and PI3K/AKT/mTOR inhibitors show clinical benefit in selected biomarker populations.
ctDNA monitoring can detect resistance and guide therapeutic escalation in real time.
Abstract
Breast cancer is the most common cancer in women worldwide, and most cases are oestrogen receptor (ER) positive. These cancers are usually treated with hormone (endocrine) therapy and targeted drugs, which have greatly improved survival outcomes. However, many patients eventually stop responding to treatment, as the cancer develops resistance. Research has shown that ER-positive breast cancer is not one single disease but rather a group of subtypes driven by different genetic changes. New technologies, such as next-generation sequencing and blood tests that detect tumour DNA (ctDNA), allow for the identification of genetic differences. This can help guide more personalised treatment decisions. Promising new therapies include oral selective oestrogen receptor degraders and drugs targeting growth pathways such as PI3K/AKT/mTOR inhibitors. Wider access to molecular testing and ongoing drug…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
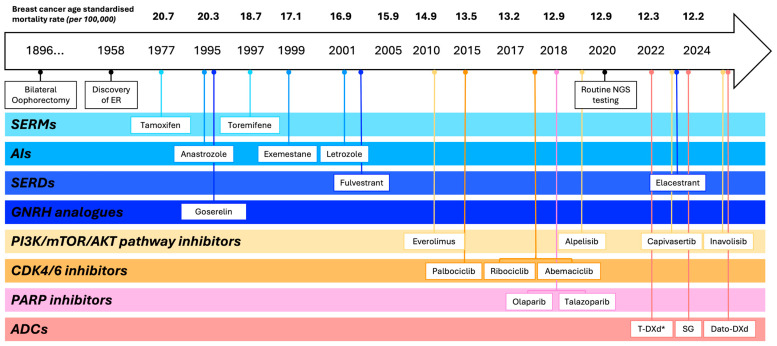 Figure 1
Figure 1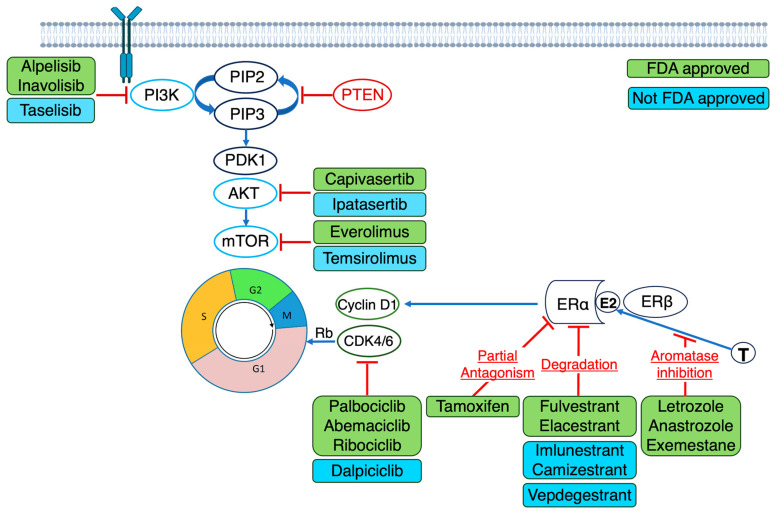 Figure 2
Figure 2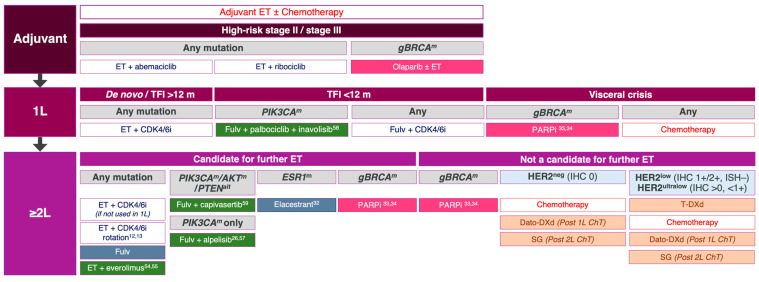 Figure 3
Figure 3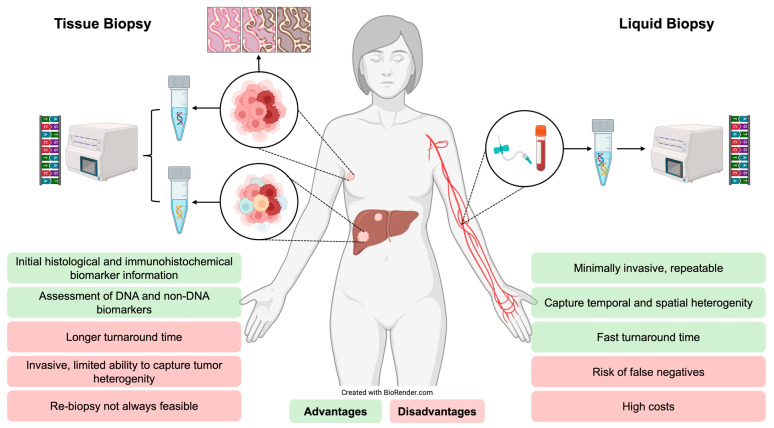 Figure 4
Figure 4- —National Breast Cancer Endowed Chair 17-02
- —White Walker Scholarship, UNSW
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer Genomics and Diagnostics · Cancer Cells and Metastasis · Breast Cancer Treatment Studies
1. Introduction
Globally, breast cancer is the most commonly diagnosed malignancy and the leading cause of cancer-related mortality among women [1,2]. Up to ~30% of high-risk early-stage patients develop metastatic disease, with risk persisting for decades; in ER-positive breast cancer, recurrences can occur up to twenty years following diagnosis, with nearly half occurring beyond five years post-endocrine therapy [3,4]. Survival rates vary widely among patients with a similar stage and subtype, complicating risk stratification and treatment decisions [1]. Expanding knowledge of the molecular heterogeneity of breast cancer has advanced precision oncology beyond conventional oestrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor 2 (HER2) subtyping toward therapies targeting tumour-specific molecular alterations to improve clinical outcomes [5]. This review integrates advancements in molecular phenotyping with practical treatment algorithms for advanced ER-positive breast cancer, providing a clinically focused framework that bridges emerging genomic insights with therapeutic decision making.
2. The Molecular Landscape of Advanced ER-Positive Breast Cancer
Breast oncology was among the earliest fields to adopt targeted therapy, beginning with surgical oophorectomy [6,7] followed by the introduction of anti-oestrogenic agents for ER-positive disease and the identification of HER2 amplification as a therapeutic target [8]. Since then, the assessment of ER, PR and HER2 remains central for subtyping and guiding initial therapeutic decisions [9,10]. More recently, the incorporation of cyclin-dependent kinase 4/6 (CDK4/6) inhibitors to endocrine therapy has become a cornerstone treatment in both high-risk early-stage and advanced ER-positive breast cancer, significantly improving survival. The sequential evolution of hormone and targeted therapies for ER-positive breast cancer (Figure 1) has contributed to a sustained decline in breast cancer-related mortality [11,12]. However, resistance remains inevitable, highlighting the need for novel treatment strategies. Trials such as MAINTAIN and postMONARCH have evaluated the continuation or rechallenge of CDK4/6 inhibitors in selected patients, showing modest improvements in progression-free survival (PFS), particularly after a prolonged response to CDK4/6 inhibition [13,14].
Advances in molecular profiling have revealed key truncal and acquired alterations in ER-positive breast cancer. Frequently observed alterations include mutations in ESR1 (10–40%), TP53 (10–34%), PIK3CA (30%), CCND1 amplifications (9–22%), GATA3 (12–20%), MAP3K1 (8–10%), FGFR1 amplifications (10–15%), CDH1 (9%), PTEN (7%), AKT1 (7%), germline BRCA1/2 (4%) and germline PALB2 (1%) [15,16,17]. These mutations drive diverse biological processes including endocrine resistance, PI3K/AKT/mTOR signalling, cell cycle regulation and chromatin remodelling, influencing both prognosis and therapeutic response [15,16,17]. Alterations such as PIK3CA and ESR1 are actionable, with approved or emerging therapies, while others such as TP53 and GATA3 are primarily prognostic or mechanistic [15,16,17]. Figure 2 outlines key signalling pathways and corresponding targeted therapies supported by phase III trial data.
International guidelines recommend comprehensive genomic profiling, either tumour or circulating tumour DNA (ctDNA), in advanced ER-positive breast cancer [23,24,25]. The American Society of Clinical Oncology (ASCO) supports utility beyond the first-line setting to influence therapeutic decision making [23]. In 2020, the European Society for Medical Oncology (ESMO) updated guidelines to recommend routine testing when results are likely to influence treatment selection, guided by the ESMO Scale for Clinical Actionability of Molecular Targets (ESCAT) framework, which stratifies molecular alterations by strength of clinical evidence [24,26]. Table 1 summarises clinically relevant alterations in ER-positive breast cancer according to ESCAT tiers [24,26].
Mutations in the ligand-binding domain of the ESR1 gene, which encodes ERα, represent a well-established mechanism of acquired endocrine resistance [36]. These mutations result in constitutive ERα activation, driving oestrogen-independent growth [10,36]. The most common ESR1^mut^, along with their prevalence and clinical implications, are summarised in Table 2. Their frequency increases with longer exposure to endocrine therapy and is associated with poorer results, with outcomes varying depending on the specific ESR1^mut^ and the presence of dual mutations [36,37,38]. The most prevalent ESR1^mut^ include D538G and Y537S; other variants include Y537N, Y537C, L536H, L536P, L536R, S463P and E380Q [36].
The PIK3CA gene has emerged as a clinically relevant biomarker in advanced ER-positive breast cancer [18,40]. Activating mutations in PIK3CA result in hyperactivation of the PI3K/AKT/mTOR pathway, promoting oestrogen-independent growth [10,40]. These mutations are among the most common in advanced ER-positive breast cancer, occurring in 28–46% of cases, and are typically truncal, present in the primary tumour at diagnosis [40,41]. Discordance of PIK3CA^mut^ between the primary tumour and metastatic sites is infrequent at 9.8% (95% CI, 7–13%) [41]. PIK3CA^mut^ are associated with poor prognosis (HR 1.2, 95% CI 0.9–1.5 and p < 0.001), with a meta-analysis demonstrating an 8-month OS difference, and chemotherapy resistance [10,40,42]. The most frequent PIK3CA^mut^ include H1047R (35%), E545K (17%) and E542K (10%), with biochemical differences, though current evidence does not link them to differential clinical outcomes [18,43]. A subset of tumours harbour multiple PIK3CA^mut^, possibly reflecting clonal evolution and greater pathway activation, though prognostic implications remain uncertain [43]. Co-occurring PIK3CA^mut^ and ESR1^mut^, observed in 10–15% of endocrine-resistant cases, may confer synergistic resistance [44,45]. Other PI3K pathway alterations, including AKT1^mut^ and PTEN^del^, each with an estimated incidence of 6%, are implicated in endocrine resistance and represent emerging therapeutic targets [5,10,26].
Additional clinically actionable alterations include BRCA1/2^mut^, which are present in ~4–5% of ER-positive breast cancers, the majority of which (~75%) are germline [10]. BRCA1/2^mut^ tumours exhibit defective homologous recombination repair and are sensitive to poly-(ADP-ribose)-polymerase (PARP) inhibitors, offering a therapeutic option in early and advanced disease. Although associated with genomic instability and aggressive biology, the prognostic impact of BRCA1/2^mut^ varies across studies, and sensitivity to PARP inhibitors may provide an important counterbalance to their adverse biological features [30,31]. Similarly, germline PALB2^mut^, occurring in 1–2% of ER-positive breast cancers, impair homologous recombination repair and confer comparable sensitivity to PARP inhibitors [26].
Somatic HER2^mut^, distinct from HER2 amplification, occur in 3–6% of ER-positive ductal and 18–26% of pleomorphic lobular carcinomas [10,46]. These mutations promote endocrine resistance through ER-HER2 crosstalk and are associated with shorter PFS and reduced endocrine sensitivity [10,47]. With the emergence of molecular targets, the contemporary management of advanced ER-positive breast cancer (Figure 3) has shifted to prioritise biomarker-directed therapies over chemotherapy or antibody–drug conjugates (ADCs) when actionable targets are identified.
3. Molecular Profiling in Advanced ER-Positive Breast Cancer
Molecular profiling plays a central role in advanced ER-positive breast cancer, supporting prognostication, real-time monitoring of therapeutic response and identification of actionable alterations for targeted therapy [10,48]. It can be performed using tumour tissue or ctDNA from blood, referred to as a liquid biopsy [8]. While tissue biopsy remains the gold standard for initial diagnosis and immunohistochemical (IHC) profiling of ER, PR and HER2, it may inadequately capture spatial and temporal tumoural heterogeneity [10]. Distinct metastases can harbour different mutations, and resistance alterations can emerge under therapeutic pressure [48,49]. Up to 40% of tumours switch molecular subtype upon progression, which has prompted guideline recommendations to consider repeat biopsy where feasible [10,25]. Subtype switching with loss of ER expression occurs in 10–20% of cases, whilst HER2 status changes in 5–15%, most frequently as HER2 gain [50,51,52]. Such changes are clinically relevant as they may confer resistance to endocrine therapy, alter prognosis or open eligibility for targeted agents, such as HER2-directed therapies in cases of acquired HER2 expression. However, repeat tissue sampling is not always feasible, as is limited by procedural risks, anatomical inaccessibility and patient quality-of-life implications [10,48].
In this context, plasma-based ctDNA profiling offers a minimally invasive alternative that captures tumour heterogeneity and molecular evolution [48]. Cell-free DNA (cfDNA) is mainly released via apoptosis and necrosis, a proportion of which is derived from tumour cells (ctDNA) [10,49]. The ctDNA fraction varies from 0.01 to 0.1% in early-stage disease to 5 to 10% in advanced disease, influenced by tumour burden, proliferative rate and breast cancer subtype [10,49]. Detection methods include digital droplet polymerase chain reaction (ddPCR) for precise detection of known mutations or next-generation sequencing (NGS) for broad multi-gene profiling [10,37,49]. Liquid biopsy enables repeat, relatively non-invasive and real-time assessment and quantification of genomic alterations to capture intra-tumoural heterogeneity and clonal evolution, although it can be limited by false-negative results from low-shedding tumours [10,48,53]. Figure 4 summarises the advantages and limitations of tissue versus liquid biopsy for molecular profiling in ER-positive breast cancer.
Retrospective studies report ~60% concordance between tissue- and plasma-based molecular profiling, with ~20% of variants unique to either source [48]. ESR1^mut^ are the most frequent mutations exclusive to ctDNA (55%), while PIK3CA^mut^ demonstrated the highest concordance (70%) [48]. The prospective plasmaMATCH trial reported 93% sensitivity for ctDNA detection of ESR1, PIK3CA, HER2 and AKT1 mutations compared to tissue sequencing, which increased to 98% with contemporaneous sampling [54]. Meta-analyses confirm high sensitivity and specificity for ctDNA detection of ESR1^mut^ (sensitivity 75.5%; specificity 88.2%) and PIK3CA^mut^ (sensitivity 73%; specificity 83%) [55,56]. Dynamic ctDNA monitoring has been proposed as a surrogate biomarker of treatment efficacy. In MONALEESA-3, ctDNA changes between cycles 1 and 4 correlated strongly with PFS (HR 0.29, 95% CI 0.22–0.38 and p < 0.0001) and OS (HR 0.23, 95% CI 0.17–0.31 and p < 0.0001) [57].
Together, these findings support ctDNA as a valuable tool for molecular profiling in advanced ER-positive breast cancer, although tissue biopsy remains critical for initial diagnosis and IHC assessment [54].
4. Therapies Targeting Genomic Aberrations in Advanced ER-Positive Breast Cancer
Genomic profiling plays a critical role in identifying targetable alterations in advanced ER-positive breast cancer, offering the potential to improve clinical outcomes [48]. Several mutations are classified as ESMO ESCAT tier I or II, denoting readiness for clinical use or promising investigational therapies (Table 1) [26]. The identification of tumour-specific oncogenic driver mutations has triggered a surge in drug development and reshaped the clinical trial landscape (Figure 3).
4.1. ESR1 Mutations
New therapies that degrade ERs have been developed to retain clinical activity in ESR1^mut^ by targeting both mutant and wild-type ERα, unlike conventional endocrine therapies such as AIs [10,58]. The phase III SoFEA and EFECT trials demonstrated that ESR1^mut^ predicted poor response to AIs, with improved outcomes using fulvestrant, a first-generation intramuscular selective ER degrader (SERD) (median PFS 2.4 vs. 3.9 months; HR 0.59, p = 0.01) [58]. However, the efficacy of fulvestrant has been limited by poor bioavailability and limited dosing [10,46].
Elacestrant, a next-generation oral SERD, received FDA approval based on the phase III EMERALD trial [9,10,28]. Elacestrant significantly improved PFS compared to standard endocrine monotherapy in patients who progressed with endocrine therapy and a CDK4/6 inhibitor (HR 0.70, 95% CI 0.55–0.88 and p = 0.002), with greater benefit observed in ESR1^mut^ tumours (HR 0.55, 95% CI 0.39–0.77 and p = 0.0005) [28]. In a post hoc analysis, a longer duration of prior CDK4/6 inhibitor (>12 months) was predicted for superior PFS of 8.6 months in those receiving elacestrant vs. 1.9 months for those receiving standard endocrine therapy (HR 0.41, 95% CI 0.26–0.63 and p = 0.014) [59]. These results informed FDA approval criteria and highlight the importance of integrating molecular characteristics and functional response to prior therapy to optimise patient selection for second-line endocrine monotherapy.
The phase II SERENA-2 trial evaluated camizestrant, an oral SERD, in the second-line setting, demonstrating superior PFS compared with fulvestrant in the overall population (7.2 vs. 3.7 months; HR 0.59, 90% CI 0.42–0.82 and p = 0.0170) [60]. This correlated with early reductions of ctDNA ESR1 variant allele frequency (VAF) by cycle 2 [60]. Other oral SERDs investigated in the second-line setting include imlunestrant (phase III EMBER-3 trial) and giredestrant (phase II acelERA trial), both demonstrating benefits limited to the ESR1^mut^ population [61,62].
In the phase III VERITAC trial, vepdegestrant, an ER proteolytic-targeting chimera (PROTAC) degrader, demonstrated superior efficacy to fulvestrant in the ESR1^mut^ population (PFS 5.0 vs. 2.1 months; HR 0.58, p < 0.001) [63].
Given the benefit of oral SERDs in advanced disease, several phase III trials (LiDERA (NCT04961996), EMBER-4 (NCT05514054) and ELEGANT (NCT06492616)) are investigating switching from AIs to SERDs in high-risk early breast cancer. Table 3 summarises completed and ongoing phase II and III trials investigating novel endocrine therapies stratified by ESR1^mut^ status.
Serial ctDNA monitoring has detected emerging ESR1^mut^ at a median of 6.7 months before radiographic progression [46]. The phase III PADA-1 and SERENA-6 trials enrolled patients on first-line AI plus CDK4/6 inhibitor therapy with rising ESR1^mut^ on ctDNA (by NGS) without radiographic progression [29,68]. Patients were randomised to continue their current therapy or switch the endocrine therapy backbone to fulvestrant (PADA-1) or camizestrant (SERENA-6) while continuing their CDK4/6 inhibitor [29,68]. Both trials demonstrated improved PFS with early switching (PADA-1: 11.9 vs. 5.7 months, HR 0.61, 95% CI 0.43–0.86 and p = 0.004; SERENA-6: 16.0 vs. 9.2 months; HR 0.44, 95% CI 0.31–0.60 and p < 0.00001) [29,68]. Patients were monitored with ctDNA testing every 2–3 months; however, only 10–17% of patients were randomised to escalation of therapy, making this approach resource intensive and cost prohibitive for routine implementation into clinical practice [29,68].
Ongoing trials such as persevERA and SERENA-4 are evaluating whether upfront SERDs can prevent ESR1^mut^-driven resistance, which, if positive, may represent an alternative strategy, avoiding the need for serial ctDNA testing [69,70].
4.2. Alterations in PIK3CA, AKT and PTEN
The PI3K/AKT signalling pathway plays a critical role in various physiological processes, including cell growth, proliferation, survival and the regulation of glucose and lipid metabolism [72]. Consequently, therapies targeting this pathway have a narrow therapeutic index, and tolerability has proven clinically challenging.
Everolimus, an mTOR inhibitor, was the first PI3K/AKT pathway-directed therapy approved in breast cancer. The phase III BOLERO-2 trial demonstrated a PFS benefit of 10.6 months with exemestane plus everolimus vs. 4.1 months with exemestane alone (HR 0.36, 95% CI 0.27–0.47, p < 0.001) in patients who had progressed on endocrine therapy [38]. The phase II PrE0102 study similarly demonstrated a PFS benefit of second-line fulvestrant plus everolimus of 10.3 months vs. 5.1 months with fulvestrant monotherapy (HR 0.61, 95% CI 0.40–0.92 and p = 0.02) [73]. Both trials predated routine NGS profiling and CDK4/6 inhibitor use, therefore limiting applicability in the current clinical landscape. A recent single-arm study evaluating fulvestrant plus everolimus post-CDK4/6 progression demonstrated a median PFS of 6.8 months and validated ctDNA dynamics as a prognostic biomarker [74].
Alpelisib, an oral α-selective PIK3CA inhibitor, demonstrated efficacy in the phase III SOLAR-1 trial when combined with fulvestrant versus fulvestrant monotherapy in the second-line setting for advanced ER-positive/HER2-negative breast cancer [18]. This combination achieved a 45% reduction in the risk of progression and a 7.9-month improvement in OS, although it did not reach the pre-specified threshold for statistical significance [18]. However, 90% of patients in SOLAR-1 had not received a CDK4/6 inhibitor prior, which is now standard first-line therapy [18]. The phase II BYLieve trial, assessing alpelisib post-progression on a CDK4/6 inhibitor, demonstrated a median PFS of 7.5 months and an OS ranging from 20.7 to 29.0 months [19]. Based on these results, alpelisib received FDA approval for patients with advanced PIK3CA^mut^ ER-positive/HER2-negative breast cancer. However, high toxicity rates including grade ≥ 3 hyperglycaemia (36%), grade ≥ 3 rash (10%), 25% discontinuation rate and 64% dose reductions and/or interruptions have limited clinical utility [18,19]. Hyperglycaemia is the most frequent adverse event, impacting up to 60% of patients receiving alpelisib, and safety in patients with type 1 or 2 diabetes has not been established. Use of prophylactic metformin has been shown to reduce the incidence and severity of alpelisib-induced hyperglycaemia, any grade (44.1%) and grades 3–4 (5.9%), in the METALLICA study [75], facilitating continuation of therapy.
Next-generation mutant selective PI3Kα degraders, such as inavolisib, aim to spare wild-type PI3K signalling, thereby reducing off-target toxicities [20]. The phase III INAVO120 trial evaluated inavolisib or placebo with fulvestrant and palbociclib in the first-line metastatic PIK3CA^mut^ patients who relapsed on or within 12 months of adjuvant endocrine therapy [20]. Inavolisib significantly improved PFS (15 vs. 7.3 months; HR 0.43, 95% CI 0.32–0.59 and p < 0.001) and OS (34.0 vs. 27.0 months; HR 0.67, p = 0.019), marking the first PIK3CA pathway therapy to demonstrate an OS benefit in breast cancer and underscoring its potential to change first-line treatment standards [20]. Inavolisib was also more tolerable when compared to alpelisib, with grade ≥ 3 hyperglycaemia (6%), grade ≥ 3 rash (2.5%) and a 7% discontinuation rate [20]. This study has shifted the mutation testing paradigm to before first-line metastatic therapy for some patients. Early-phase trials investigating novel, mutant-specific PIK3CA inhibitors such as RLY-2608 (NCT05216432) and STX-478 (NCT05768139) have demonstrated promising efficacy and tolerability and are now progressing into phase III evaluation.
The phase III CAPItello-291 trial evaluated capivasertib, a pan-AKT inhibitor, plus fulvestrant vs. fulvestrant monotherapy in the second-line setting for advanced ER-positive/HER2-negative breast cancer, regardless of but stratified by mutational status, with 41% of patients harbouring an AKT pathway alteration (defined as PIK3CA^mut^, AKT^mut^ and/or PTEN^del^) [21]. Capivasertib significantly prolonged PFS in the intention-to-treat (ITT) population (7.2 vs. 3.6 months; HR 0.60, 95% CI 0.51–0.71 and p < 0.001) and the pathway-altered population (7.2 vs. 3.6 months; HR 0.50, 95% CI 0.32–0.59 and p < 0.001) [21]. Despite similar efficacy, FDA approval was granted only for patients with an identified pathway alteration. Capivasertib was relatively well tolerated with grade ≥ 3 hyperglycaemia (2%), grade ≥ 3 rash (12%) and a 13% discontinuation rate [21]. A phase III study of capivasertib plus fulvestrant and a CDK4/6 inhibitor in the first-line metastatic setting is currently underway (NCT04862663). The phase III FINER trial assessed ipatasertib, an alternative pan-AKT inhibitor, plus fulvestrant in the second-line setting, stratified by AKT pathway alterations [76]. Ipatasertib improved PFS in the ITT population (5.3 vs. 1.9 months; HR 0.61, 95% CI 0.46–0.81 and p = 0.0007), with greater benefit in the mutant cohort (5.5 vs. 1.9 months; HR 0.47, 95% CI 0.31–0.72 and p = 0.0005) [76].
Table 4 summarises completed and ongoing phase II and III trials investigating therapies targeting the PIK3CA/PTEN/AKT pathway in advanced breast cancer.
4.3. BRCA1/2 and PALB2 Mutations
The benefit of PARP inhibitors for patients with germline BRCA1/2^mut^ is well established, supported by phase III trials OlympiAD and EMBRACA [30,31]. OlympiAD demonstrated improved PFS with olaparib vs. chemotherapy (7.0 vs. 4.2 months; HR 0.58, p < 0.001) in advanced HER2-negative breast cancer (~50% ER positive) [30]. EMBRACA demonstrated superior PFS with talazoparib (PFS 8.6 vs. 5.6 months; HR 0.54, p < 0.001) [31]. While the efficacy of PARP inhibitors in somatic BRCA1/2^mut^ remains under investigation, a phase II olaparib trial suggested comparable activity, with an overall response rate (ORR) of 50% [32]. This trial also demonstrated benefit from olaparib in patients with a germline PALB2^mut^ (ORR 82%) [32]. Ongoing studies are investigating PARP inhibitors in earlier treatment settings and novel combinations, including with oral SERDs, as in the EvoPAR-Breast01 trial (NCT06380751).
4.4. HER2 Mutations
Trials specific to HER2^mut^ breast tumours are limited due to their rarity, with most evidence arising from pan-tumour basket trials. While FDA-approved therapies exist for ER-positive/HER2-non-amplified breast cancer based on HER2 IHC expression, no FDA-approved therapies currently cover HER2^mut^ breast cancers specifically. Furthermore, in practice, HER2^mut^ testing is not yet routine, as it requires NGS panels rather than standard IHC or in situ hybridisation (ISH) testing.
The phase II SUMMIT trial evaluated neratinib, a pan-HER tyrosine kinase (TKI), as monotherapy or combined with fulvestrant—with or without trastuzumab—in patients with advanced HER2^mut^ ER+/HER2-negative breast cancer who had progressed on prior CDK4/6 inhibitor [33]. The triplet regimen achieved an ORR of 39% and a median PFS of 8.3 months [33]. Trastuzumab deruxtecan (T-DXd), an ADC consisting of a HER-2 targeted monoclonal antibody combined via a cleavable peptide linker to a topoisomerase I inhibitor payload, has demonstrated durable responses in heavily pre-treated HER2^mut^ non-small-cell lung cancer (NSCLC) in the DESTINY-Lung01 trial, leading to FDA approval [79]. In the DESTINY-Pantumour01 basket trial, T-DXd demonstrated a promising ORR of 50% in the HER2^mut^ breast cancer cohort [34]. A phase I trial (NCT05372614) is currently evaluating the combination of T-DXd with Neratinib in patients with HER2 amplification or HER2^mut^ [80].
5. Current Limitations and Future Directions of a Molecular Phenotypic Approach to Treating ER-Positive Breast Cancer
Despite growing clinical evidence and support from international guidelines, several key limitations hinder the widespread integration of molecular profiling in advanced ER-positive breast cancer. High upfront costs of NGS panels, limited reimbursement pathways and variation in test availability restrict patient access. Furthermore, access to matched targeted therapies remains largely confined to clinical trials or cost-share programs, further limiting accessibility, particularly in under-resourced settings.
Implementing molecular profiling requires specialised laboratory infrastructure, bioinformatics support and clinical expertise to interpret results. Tissue-based NGS is more established but often involves delays, limiting utility in time-sensitive scenarios, and may require repeat biopsies, which are not always feasible or acceptable. ctDNA offers a minimally invasive, real-time alternative, though sensitivity is limited in early-stage, low-volume or ER-positive subtypes. ddPCR is a cost-effective, highly sensitive method for detecting known mutations, but it has not been widely used in pivotal phase III clinical trials; thus, comparative studies would be required to facilitate widespread expansion of this technology. Effective molecular profiling requires a nuance understanding of the technical limitations and the complex, non-binary implications of genomic data to provide personalised treatment recommendations.
To address these challenges, health systems are increasingly adopting centralised testing models, multidisciplinary molecular tumour boards and partnerships with reference laboratories to standardise reporting and facilitate expert input. Centralised testing can achieve economies of scale and decrease per-sample costs, while clinical trial participation may provide subsidised or no-cost testing opportunities. Ultimately, demonstrating cost effectiveness of molecular profiling will be essential to support broader government or insurance reimbursement. A pragmatic strategy to improve the cost effectiveness in ER-positive breast cancer is to prioritise a focused panel testing approach limited to the most common, currently actionable molecular targets, such as ESR1, PIK3CA, AKT, PTEN, BRCA1/2 and HER2, acknowledging that a small percentage of variants may be missed with this methodology. Investment in clinician education and local expertise through collaborative partnerships with academic centres will build capacity and ensure the integration of molecular profiling into routine practice.
The use of molecular profiling in clinical care raises important ethical considerations. Informed consent processes must clearly explain the scope of testing, potential incidental findings and implications for family members. Robust safeguards for data privacy and cybersecurity are essential, as genomic information constitutes highly sensitive personal data. Clear policies on data sharing, storage and secondary use are necessary to protect patient autonomy while supporting research and innovation. Addressing these ethical challenges proactively will help ensure that precision medicine advances in a way that maintains public trust and protects patient rights.
Future molecular profiling in advanced ER-positive breast cancer aims to improve therapeutic precision by expanding actionable alterations. Beyond guiding targeted therapies, it may define endocrine therapy duration, identify late recurrence risk, personalise surveillance and adapt treatment to resistance mutations. Early evidence supports therapeutic escalation with the emergence of detectable ESR1^mut^, and future studies should explore expanding this approach to other mutational alterations that may emerge with therapy selection. For example, HER2^mut^ detection may warrant early change to HER2-directed TKIs or ADCs; somatic BRCA1/2^mut^ could support early introduction of PARP inhibitors; Retinoblastoma (Rb) loss, implicated in CDK4/6 inhibitor resistance, may justify earlier second-line therapy; and cyclin E amplification may predict benefit from early CDK2 inhibitors. Emerging CDK2 inhibitors are being developed to address resistance to CDK4/6 inhibition, particularly in the setting of cyclin E amplification or Rb loss. Several selective CDK2 inhibitors, such as PF-07104091 (NCT04553133) [81] and BLU-222 (NCT05252416) [82], are currently in phase I/II clinical trials in the second line and beyond. Early results have demonstrated acceptable tolerability and preliminary anti-tumour activity, with a disease control rate of 61.5% in one cohort [81], supporting ongoing development.
Looking ahead, integrating ctDNA kinetics with clinical features and artificial intelligence driven analytics could enhance real-time treatment decisions, enabling therapy escalation or de-escalation based on tumour evolution and early detection of resistance. These dynamic risk models have the potential to refine risk stratification, optimise sequencing of targeted therapies and improve clinical outcomes for patients with advanced ER-positive breast cancer. However, prospective validation is required to confirm their clinical utility and ensure safe implementation in routine practice.
Nanotechnology holds significant potential to advance precision oncology in ER-positive breast cancer, as targeted nanoparticle drug delivery can enhance the therapeutic index by concentrating treatment within tumour cells while minimising toxicity to normal tissues [83]. When integrated with molecular profiling and ctDNA monitoring, nanocarriers may enable adaptive therapy, facilitate combination drug delivery and help overcome resistance mechanisms driven by mutations such as ESR1 or PIK3CA [83].
Continued therapeutic development is essential as new treatments modify the natural history of ER-positive breast cancer and reveal novel resistance mechanisms. Most trials target ERs, cell cycle machinery and the PIK3CA/AKT pathway—often combined with an endocrine therapy backbone—highlighting the need for more effective endocrine agents. As novel combination therapies move into earlier treatment lines, newer treatment options are clearly needed upon progression. Since NGS-guided approaches focus on mutations, mutation-agnostic therapies like emerging ADCs and epigenetic modulators such as KAT2 inhibitors [84] remain important for patients without targetable mutations.
6. Conclusions
Molecular profiling is now a cornerstone to managing ER-positive advanced breast cancer, serving as a predictive and prognostic biomarker. International guidelines recommend routine implementation, using tissue or ctDNA, to identify actionable genomic alterations such as PIK3CA and ESR1, which has catalysed a surge in drug development and clinical trials, expanding treatment options for patients with advanced, incurable disease. Beyond treatment selection, molecular profiling may aid prognostication and monitoring of treatment response; however, critical unanswered questions regarding clinical validity, optimal integration into treatment pathways and impact on survival outcomes remain uncertain. Addressing real-world barriers is essential to ensure equitable access and facilitate routine implementation into clinical practice globally.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bray F. Laversanne M. Sung H. Ferlay J. Siegel R.L. Soerjomataram I. Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA Cancer J. Clin.20247422926310.3322/caac.2183438572751 · doi ↗ · pubmed ↗
- 2Kim J. Harper A. Mc Cormack V. Sung H. Houssami N. Morgan E. Mutebi M. Garvey G. Soerjomataram I. Fidler-Benaoudia M.M. Global patterns and trends in breast cancer incidence and mortality across 185 countries Nat. Med.2025311154116210.1038/s 41591-025-03502-339994475 · doi ↗ · pubmed ↗
- 3Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) Reductions in recurrence in women with early breast cancer entering clinical trials between 1990 and 2009: A pooled analysis of 155,746 women in 151 trials Lancet 20244041407141810.1016/S 0140-6736(24)01745-839396348 · doi ↗ · pubmed ↗
- 4Pan H. Gray R. Braybrooke J. Davies C. Taylor C. Mc Gale P. Peto R. Pritchard K.I. Bergh J. Dowsett M. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years N. Engl. J. Med.20173771836184610.1056/NEJ Moa 170183029117498 PMC 5734609 · doi ↗ · pubmed ↗
- 5Rassy E. Mosele M. Di Meglio A. Pistilli B. Andre F. Precision oncology in patients with breast cancer: Towards a ‘screen and characterize’ approach ESMO Open 2024910371610.1016/j.esmoop.2024.10371639303452 PMC 11439525 · doi ↗ · pubmed ↗
- 6Beatson G.T. On the Treatment of Inoperable Cases of Carcinoma of the Mamma: Suggestions for a New Method of Treatment, with Illustrative Cases Trans. Med.-Chir. Soc. Edinb.189615153179 PMC 551837829584099 · pubmed ↗
- 7Beatson G.T. The treatment of cancer of the breast by oophorectomy and thyroid extract Br. Med. J.1901211451148
- 8Shatsky R. Parker B.A. Bui N.Q. Helsten T. Schwab R.B. Boles S.G. Kurzrock R. Next-Generation Sequencing of Tissue and Circulating Tumor DNA: The UC San Diego Moores Center for Personalized Cancer Therapy Experience with Breast Malignancies Mol. Cancer Ther.2019181001101110.1158/1535-7163.MCT-17-103830926636 · doi ↗ · pubmed ↗