Selection Signatures in the Genome of Dzhalgin Merino Sheep Breed
Alexander Krivoruchko, Olesya Yatsyk, Antonina Skokova, Elena Safaryan, Ludmila Usai, Anastasia Kanibolotskaya

TL;DR
This study identifies genes in Dzhalgin Merino sheep that may be responsible for their high productivity and adaptability to harsh environments.
Contribution
The study discovers 185 genes, including key candidates, in Dzhalgin Merino sheep under selection pressure for adaptive and productive traits.
Findings
A total of 185 genes were identified in loci showing evidence of selection in Dzhalgin Merino sheep.
Seven genes (EPHA6, MLLT3, ROBO1, KIAA0753, MED31, SLC13A5, and ELAVL4) were highlighted as key candidates for further research.
The identified genes are potentially involved in growth, development, and reproduction processes.
Abstract
Selection acting on farm animal populations results in the formation of specific regions in their genomes that reflect the influence of the selection process. Studying such regions allows us to identify genes and genetic variants that play a key role in animal adaptation to environmental conditions and in the formation of productive traits. The Dzhalgin Merino breed was bred in Russia relatively recently and is characterized by high meat and wool productivity, as well as good adaptability to arid steppe conditions. Given the genetic closeness of this breed to the Australian Merino and Rambouillet, the aim of this study was to search for selection signatures and identify candidate genes at loci under selection pressure based on a comparative analysis of the Dzhalgin Merino genomes with Australian Merino and Rambouillet sheep. The identified genes are potentially involved in the formation…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
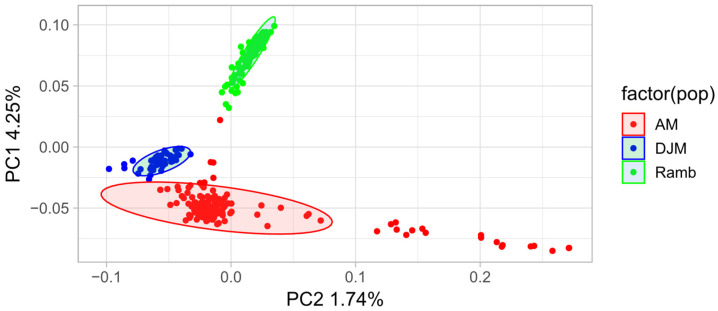 Figure 1
Figure 1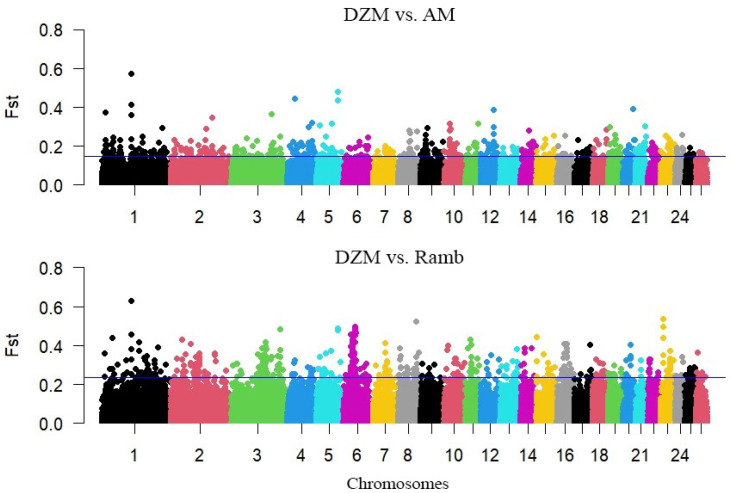 Figure 2
Figure 2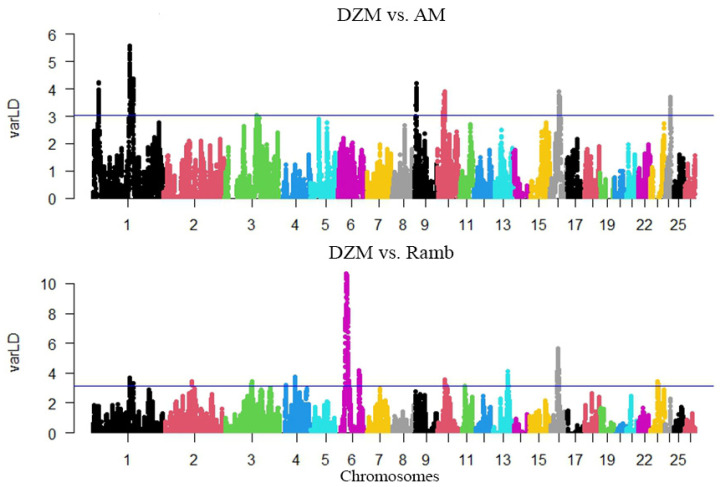 Figure 3
Figure 3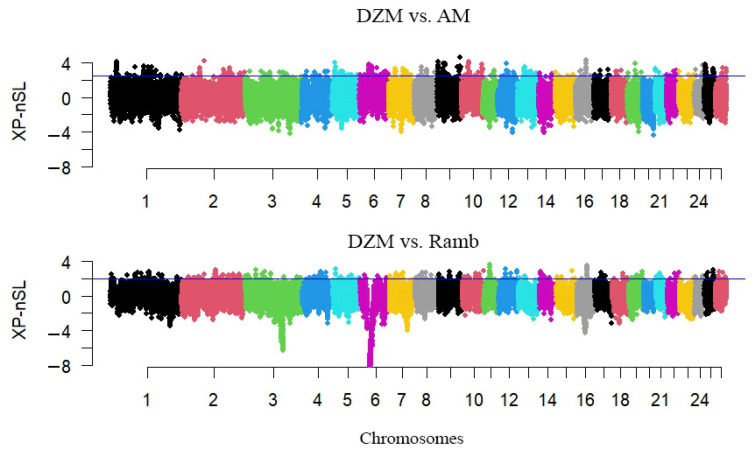 Figure 4
Figure 4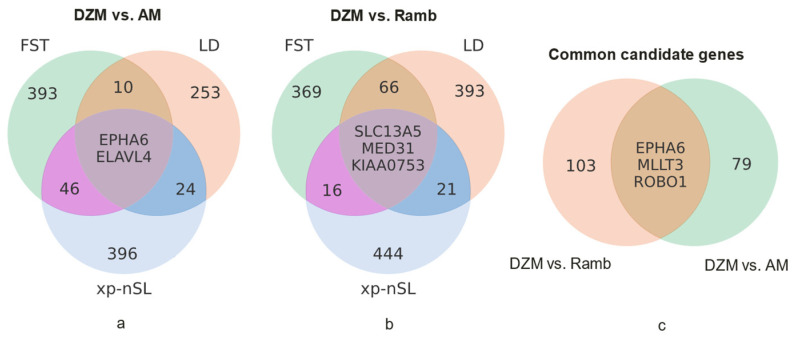 Figure 5
Figure 5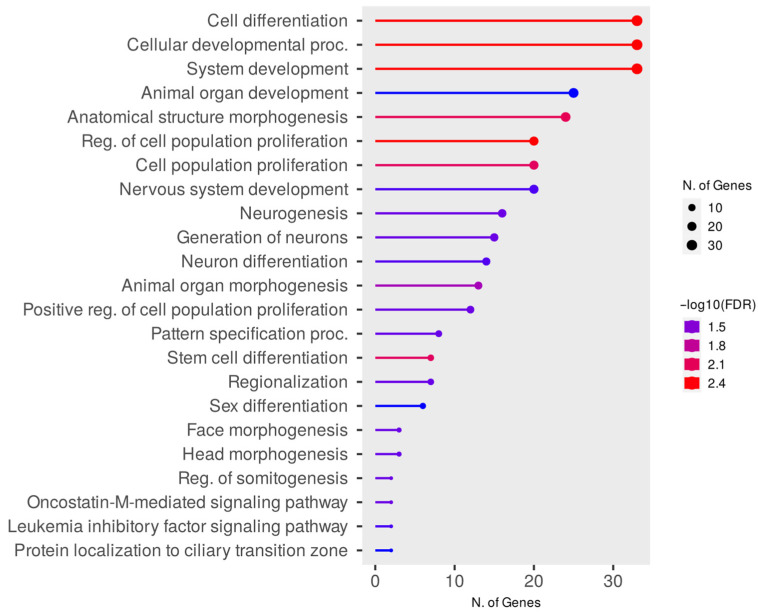 Figure 6
Figure 6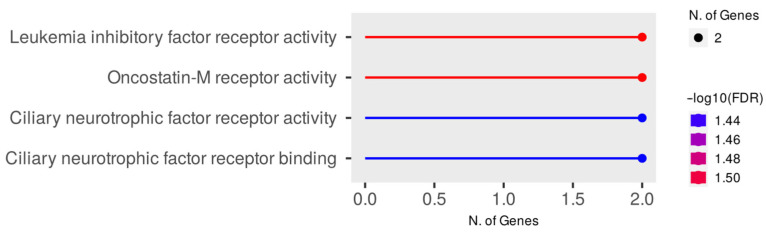 Figure 7
Figure 7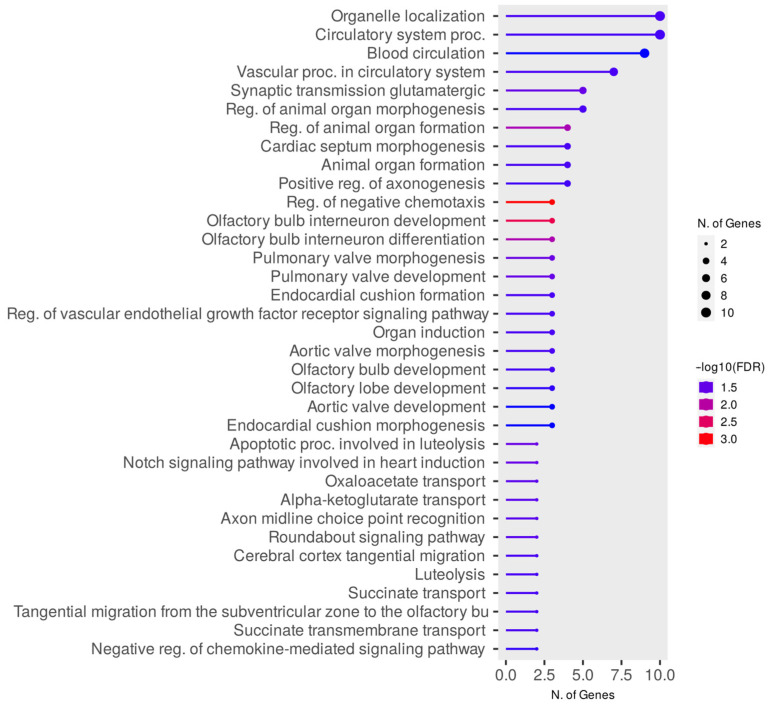 Figure 8
Figure 8- —Russian Science Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and phenotypic traits in livestock · Genetic Mapping and Diversity in Plants and Animals · Cancer-related molecular mechanisms research
1. Introduction
Positive selection acting on farm animal populations leaves characteristic imprints on their genomes, namely at loci under selection pressure. Such imprints, selection signatures, can be used to identify genes and genetic variants that play a key role in animal adaptation to specific environmental conditions or changes in their productive qualities [1]. Currently, many methods have been developed to identify genomic signatures of selection. The most reliable results are provided by an integrated approach in which loci under selection pressure are identified using several independent methods of analysis [2].
According to the literature, when conducting studies aimed at searching for selection signatures in sheep genomes, 2–3 different types of analysis are often combined. Thus, Liu Z. et al. (2022) [3] searched for selection signatures in South African Merino sheep using the FST (the fixation index), iHS (Integrated haplotype score) and xp-EHH (Cross-population extended haplotype homozygosity) methods. Among the most interesting genes located in loci under selection pressure, they identified GHR, LCORL, SMO, NCAPG, DCC, IBSP, PPARGC1A, PACRGL, PRDM5, XYLB, AHCYL2, TEFM, AFG1L and FAM184B, involved in growth processes that affect carcass characteristics and meat quality [3]. Selection signatures potentially associated with milk, wool and meat productivity, as well as resistance to parasitosis, have been identified in Slovak national breeds using ROH (Runs of homozygosity) and LD (linkage disequilibrium) methods [4]. In a study by Eydivandi S. et al. (2021) [5], simultaneous application of FST and xp-EHH methods in sheep populations from the Middle East and Europe identified genomic regions under selection pressure containing the genes CIDEA, HHATL, MGST1, FADS1, RTL1 and DGKG. Both FST and xp-EHH approaches identified 60 common genes as selection signatures, including four candidate genes (NT5E, ADA2, C8A and C8B) that were enriched for two significant Gene Ontology (GO) terms related to the adenosine metabolism pathway [5]. Identification of selection signatures in Indian sheep breeds was carried out using ROH and iHS. The results revealed selection signatures in 37 genomic regions mapping to 188 genes, including the cold adaptation gene TRPM8, meat quality traits related genes JADE2, PLEKHB2, SPP2, TSHR and UBE2B and fertility-related gene PPP3CA [6]. The use of xp-nSL (cross-population number of segregating sites by length) methods in combination with ROH in Chinese sheep breeds to identify selection signatures allowed the identification of loci associated with domestication and reproduction (TSHR, GTF2A1, KITLG, FETUB, HNRNPA1, DCUN1D1, HRG) [7].
Genomic selection signatures in Russian sheep breeds have not been extensively investigated. A study devoted to identifying selection traits associated with acclimatization and economically important traits in 15 Russian sheep breeds was conducted by Yurchenko, A.A and co-authors [8]. The study analyzed the genomes of the Buubey, Lezgin, Karachay, Karakul, Tuvan, Edilbaevskaya, Romanov, Russian Long-Wool, Altai Mountain, Grozny, Salskaya, Volgograd, Krasnoyarsk, Baikal, and Kulunda sheep breeds. The study identified genomic regions that are presumably under selection pressure. These regions contain known candidate genes associated with morphology, adaptation, and domestication (KITLG, KIT, MITF, and MC1R), wool productivity (DSG2, DSC2, and KRT2), reproduction (CMTM6, HTRA1, GNAQ, UBQLN1, and IFT88), milk production (ABCG2, SPP1, ACSS1, and ACSS2), etc. [8].
Studying the sheep genome to detect selection signatures is a promising area of scientific research, since the results obtained not only help to understand the genetic basis of adaptation, but also provide an idea of the genetic architecture of phenotypic traits and can be used to improve breeding programs. Historically, the bulk of the sheep population in Russia is represented by fine-fleeced breeds. In this regard, the search for selection signatures in merino sheep is of particular interest. Of great interest is the search for selection signatures in breeds bred for use in special climatic and natural conditions. In this case, it is possible to identify genomic loci associated with adaptation to temperature, nutrition and maintenance. One of such breeds is the Dzhalgin Merino. The breed was bred in the Stavropol Territory of the Russian Federation in 2013. It is distinguished by high rates of meat and wool productivity, good adaptability to the conditions of dry steppes. The main method of creating the breed is reproductive crossing. The mother stock for breeding the Dzhalgin Merino sheep was the Stavropol breed, and the paternal form was the Australian Merino and Caucasian breeds. Importantly, the Stavropol breed itself has a composite origin, as it was developed by crossing Mazaev and Novokavkaz ewes with American Rambouillet and Australian Merino rams, with the simultaneous use of Novokavkaz and Mazaev rams [9]. Thus, Dzhalgin Merino is genetically related to Australian Merino both directly through paternal ancestry and through the Stavropol breed, and to Rambouillet through the Stavropol breed. Given the genetic relatedness of Dzhalgin Merino sheep to both Australian Merino and Rambouillet breeds, investigating their genetic differentiation and identifying interpopulation selection signatures is of particular interest.
The aim of our study was to search for selection signatures and identify candidate genes in loci under selection pressure, potentially playing an important role in the adaptive and productive qualities of Dzhalgin Merino sheep.
2. Materials and Methods
2.1. Ethics Statement
The sample collection and study purpose were approved by the Institutional Animal Care and Use Committee (approval number 2021–0047, 10 November 2021) of the All-Russian Research Institute of Sheep and Goat Breeding, Stavropol, Russian Federation.
2.2. Experimental Animals and Sample Collection
A total of 293 animals were included in the analysis as the object of the study, all of which underwent whole-genome SNP genotyping. The sample comprised Dzhalgin Merino (DZM, n = 53), Australian Merino (AM, n = 138), and Rambouillet (Ramb, n = 102).
Whole-genome SNP genotyping data for Dzhalgin Merino were generated within the framework of this study, while the data for Australian Merino and Rambouillet were obtained from the SheepHapMap project, where animals had been genotyped using the Illumina OvineSNP50 BeadChip (Illumina, San Diego, CA, USA). The group of Australian Merino included Australian Merino (n = 50) and Australian Industry Merino (n = 88). The genotyping data of the SheepHapMap project [10] were obtained from the Web-Interfaced next-generation Database dedicated to genetic Diversity Exploration (http://widde.toulouse.inra.fr, accessed on 28 September 2025).
2.3. Genotyping and Genotyping Quality Control
Genotyping of Dzhalgin Merino animals was performed using Ovine Infinium HD BeadChip 600 K (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol. Primary processing of genotyping results was performed using Genome Studio 2.0 software (Illumina, San Diego, CA, USA).
The quality of our own genotyping data was controlled using PLINK V.1.09 software [9]. Samples in which the number of lost genotypes did not exceed 10% were included in the processing. For further analysis, we used only autosomal biallelic SNPs. SNPs without chromosomal or physical localization, SNPs with a frequency of lost genotypes exceeding 10%, and those that exceeded the threshold of deviation from Hardy–Weinberg equilibrium (HWE) p = 0.000001 were excluded. Since genotyping of the analyzed sheep breeds was performed using chips of different densities (50 k and 600 k), only polymorphisms present on both chip types were used when forming the combined dataset. Substitution positions were updated in accordance with the ARS-UI_Ramb_v2.0 genome assembly using the PLINK v1.09 program [11]. The combined dataset was subjected to quality control according to the same criteria described above. The final dataset contained data on 293 samples and 38,804 SNPs.
2.4. Principal Component Analysis
For the principal component analysis we used 36,615 SNPs, after linkage disequilibrium pruning (window size 50 SNPs, step 5 SNPs, threshold r^2^ > 0.5), performed using the PLINK V.1.09 software [11]. Principal component analysis was performed in the R v. 4.3.2 environment using the SNPRelate package [12].
2.5. Search for Selection Signatures
Calculation of the Weir and Cockerham weighted FST values for pairwise comparison of the three analyzed groups was performed using VCFtools v0.1.16 software [13], with a set window size of 100 kb and step size of 10 kb. Signals included in the upper 1% of the weighted FST values were considered as signs of selection between the two breeds.
VarLD v.1.0 software was used to assess differences in linkage disequilibrium patterns between the analyzed groups [14]. LD values calculated for sliding windows containing 50 SNPs with minor allele frequency above 5% within each autosome were transformed into standardized varLD scores, from which signatures of selection were identified. Calculations of standardized values for varLD scores, as well as visualization, were performed in R using scripts provided by the varLD authors. Signals in the top 1% of standardized scores were considered as signatures of selection between the two breeds.
xp-nSL statistics were calculated using Selscan v.2.0 with default parameters [15]. The required haplotypes were obtained by phasing using the SHAPEIT v.4.1.3 software [16]. The xp-nSL analysis results were normalized using the norm v 1.3.0 software. Positive XP-nSL values indicated the presence of hard or soft selection in the Dzhalgin Merino population, while negative values indicated the presence of hard or soft selection in the comparison populations. Since we were primarily interested in the selection features in the Dzhalgin Merino group, the signals included in the upper 1% of positive normalized xp-nSL estimates were considered as selection features. Annotation of genes located in loci under selection pressure was performed in windows of +/− 150,000 bp from the SNPs that showed selection features.
2.6. Gene Annotation and Construction of Gene Networks
Gene annotation was performed using the biomaRt package in the R environment [17].
Enrichment analysis was performed using the ShinyGO 0.80 platform [18]. The significance threshold for the false discovery rate (FDR) = 0.05. Gene networks were constructed using the platform https://string-db.org, accessed on 28 September 2025.
2.7. Visualization
Graphs were plotted in the R environment using the qqman, ggplot2, and VennDiagram packages.
3. Results
3.1. Genetic Differentiation of the Studied Groups Using PCA and FST Methods
According to the PCA results, Rambouillet and Dzhalgin Merino are presented as well-consolidated clusters, while AM showed greater intrabreed genetic variability compared to DZM and Ramb (Figure 1). In the PCA analysis, PC1 (the first principal component) accounted for 4.25% of the total genetic variability, separating all three analyzed groups of sheep, while PC2 (the second principal component) represented only 1.74% of the total genetic variability.
In the course of the research, the Weir and Cockerham FST fixation indices were calculated. Table 1 shows the weighted values for the analyzed populations. As expected, a low degree of differentiation was noted between all analyzed groups. Animals from the Australian Merino group were the closest to the Dzhalgin Merino. The greatest genetic distance was observed between the Rambouillet and the Australian Merino group. This is consistent with the results of the principal component analysis.
3.2. Search for Selection Signatures
Based on the calculations of the weighted FST values between the Dzhalgin Merino and Australian Merino groups, 2089 windows were identified that exceeded the 99% percentile with a value of 0.147479. The detected windows contained 451 protein-coding genes. When determining the selection features by FST between the Dzhalgin Merino and Rambouillet, the 99% percentile was 0.235915. This threshold was exceeded by 2087 windows, in which 454 protein-coding genes were identified. In both cases, the highest weighted FST value was identified for the window 1:127,080,001–127,180,000 on chromosome 1 (Figure 2). This window overlaps part of the GRIK1 gene.
Based on the calculations of the linkage disequilibrium values between the Dzhalgin and Australian Merino groups, 347 windows were identified that were included in 1% of the maximum standardized LD values. A total of 289 genes were identified in these windows. The highest LD value, which was 4.721373, was determined for the window located on chromosome 1; the coordinates of the window center were 1:147,516,958 (Figure 3). This point falls within the area of the ROBO1 gene. Based on the calculations of the linkage disequilibrium values between the Dzhalgin Merino and Rambouillet groups, 338 windows were identified that were included in 1% of the maximum standardized LD values. A total of 483 genes were identified in these windows. The maximum standardized LD value was 11.94841853 and was found for the window located on chromosome 6; the coordinates of the middle of the window are 6:37,577,437. This point is located in the intergenic space (Figure 3).
According to the calculation results of xp-nSL statistics between the Dzhalgin Merino and Australian Merino groups, the highest normalized xp-nSL value of 4.61038 was observed for the OAR9_95091169.1 substitution located on chromosome 9 (Figure 4). The detected polymorphism is located in the intergenic region. The established threshold corresponding to the 99th percentile was exceeded by 364 SNPs. 468 genes were identified in the adjacent regions.
For the Dzhalgin Merino and Rambouillet groups, the maximum normalized xp-nSL value was 3.62782 for the s45020.1 substitution located on chromosome 11 (Figure 4). This substitution is located in the intergenic region. The established threshold of the 99th percentile was exceeded by 360 SNPs. 484 genes were found in the adjacent regions.
To increase the reliability of identifying candidate genes in loci under selection pressure, we selected for further analysis only those genes that were located in genomic regions with selection signatures confirmed by at least two methods in each comparison (Figure 5). The selected genes were combined into gene networks. As a result, when comparing Dzhalgin Merino with Australian ones (the “DZM vs. AM” network), 82 candidate genes were identified (Supplementary Table S1, Supplementary Figure S1). In this case, the selection signatures in loci including the EPHA6 and ELAVL4 genes were confirmed by three methods at once. When comparing the Dzhalgin Merino with the Rambouillet (the DZM vs. Ramb network), 106 candidate genes were identified (Supplementary Table S2, Supplementary Figure S2), and in three of them (SLC13A5, MED31, KIAA0753) the presence of selection was confirmed by all three methods. Both formed networks have three common genes: EPHA6, MLLT3 and ROBO1.
3.3. Evaluation of PPI Interactions and Functional Enrichment of GO Terms
When constructing candidate gene networks that are supposedly under selection pressure using the STRING platform, due to the fact that the functions of most genes and proteins in the sheep organism are less well understood than in the human organism, gene networks were formed for both species at once. In all constructed gene networks, with a minimum required interaction score of medium confidence (0.400), significantly more interactions were found between proteins than would be expected for a random set of proteins of the same size and distribution (Table 2). Such enrichment indicates that the proteins are at least partially biologically related as a group.
To clarify the functions of the identified genes, functional enrichment analysis of gene ontology (GO) categories was performed using the ShinyGO 0.80 platform for molecular functions, biological processes, and cellular components. Since the functions and interactions of the analyzed genes in sheep are poorly understood, functional enrichment analysis was performed using human orthologs.
Significant enrichments for the DZM vs. AM network were identified for 23 terms of gene ontologies of biological processes (Figure 6, Supplementary Table S3) and 4 terms of molecular functions (Supplementary Table S4). No significant enrichments were identified for gene ontologies of cellular components. 50 genes from the DZM vs. AM network are involved in the enriched pathways of biological processes. The largest number of genes in the network are involved in the GO:0030154 cell differentiation, GO:0048731 system development, and GO:0048869 cellular developmental process pathways (FDR= 0.0038), including the EPHA6 and ELAVL4 genes. These two genes are involved in eight gene ontologies at once: GO:0048699 generation of neurons, GO:0030182 neuron differentiation, GO:0008283 cell population proliferation, GO:0022008 neurogenesis, GO:0009653 anatomical structure morphogenesis, GO:0007399 nervous system development, GO:0030154 cell differentiation, and GO:0048869 cellular developmental proc. The highest number of enriched terms included genes DMRTA2 (15), ROBO1, and MSX1 (14).
When analyzing the enrichment of the DZM vs. AM network according to the gene ontology of molecular functions, it was found that the LIFR and OSMR genes are involved in the pathways GO:0004923 leukemia inhibitory factor receptor activity, GO:0004924 oncostatin-M receptor activity, GO:0004897 ciliary neurotrophic factor receptor activity, GO:0005127 ciliary neurotrophic factor receptor binding (FDR = 0.0306–0.0365), (Figure 7).
Significant enrichments for the DZM vs. Ramb network were identified for 38 Gene Ontology terms of biological functions (Figure 8, Supplementary Table S5) and 1 term of cellular components (Supplementary Table S6). A total of 29 genes from the DZM vs. Ramb group are involved in the enriched pathways of biological processes. The ROBO1 gene is involved in 26 terms, ROBO2 is involved in 24, and SLIT2 in 20. The largest number of genes are involved in the GO:0051640 organelle localization and GO:0003013 circulatory system process pathways. The lowest FDR value (0.0009) was determined for the GO:0050923 reg. of the negative chemotaxis pathway.
According to the gene ontology of cellular components, significant enrichment was found only for the GO:0005944 phosphatidylinositol 3-kinase complex class IB pathway (FDR = 0.0411). This pathway involves the PIK3R5 and PIK3R6 genes.
4. Discussion
Knowledge of genetic diversity and genome regions subject to positive selection is one of the promising keys for effective management of genetic resources in sheep breeding. It allows identifying selection signatures associated with important economic traits. In this study, 185 genes located in loci under selection pressure and potentially affecting their adaptive and productive qualities were identified in Dzhalgin Merino sheep using three crosspopulation types of analysis. Comparative analysis of the genomes of Dzhalgin Merino with the genomes of animals of the Australian Merino group revealed 82 genes in loci under selection pressure (DZM vs. AM network), and 106 genes were identified when compared with animals of the Rambouillet group (DZM vs. Ramb network). The EPHA6, MLLT3 and ROBO1 genes were present in the results of both comparison groups, which emphasizes their breeding importance for the Dzhalgin Merino breed and makes them the most interesting for further study.
The EPHA6 gene belongs to the ephrin receptor tyrosine kinase family, which plays a key role in intercellular communication and regulation of cell migration, neurogenesis and angiogenesis [19]. As documented in previous studies, the EPHA6 gene is associated with fertility and reproductive traits in such species of farm animals as goats [20], sheep [21] and cattle [22]. In particular, in Jining grey goats, EPHA6 was identified as a differentially methylated and differentially expressed gene associated with litter size [20]. GWAS results identify the EPHA6 gene as a candidate gene associated with fertility traits in beef cattle [22] and Polish mountain sheep [21]. The EPHA6 gene has also been identified as a candidate gene associated with nervous system reactivity and temperament in cattle [23]. In addition, EPHA6 was identified as a positive selection gene in studies using ROH and his methods in Guishan goats [24]. EPHA6 is also associated with metabolic processes affecting fatty acid content and meat quality in Nelore cattle [25]. EPHA6 has also been identified using various genomic approaches as being under intense positive selection for milk production traits, particularly in Holstein cows [26]. Link between EPHA6 gene and breast meat color in chickens revealed [27]. Thus, despite the lack of precise data on the mechanisms of EPHA6 influence on the phenotype, it can be assumed that its participation in angiogenesis and neurohumoral regulation can affect the reproductive function and economically important traits of cattle, including traits of milk and meat productivity.
The ROBO1 gene is part of the SLIT-ROBO signaling pathway, which is involved in the formation and maturation of primordial follicles in the fetal ovaries, which affects the future reproductive potential of the animal. Studies have shown an association of ROBO1 with antral follicle count in Nelore and Angus heifers, making it an important candidate for fertility assessment [28]. The ROBO1 gene has also been identified as being under positive selection for wool fineness in sheep [29]. This suggests that ROBO1 contributes not only to fertility but also to other economically important traits such as wool quality.
The MLLT3 gene plays a key role in the activation of gene transcription and chromatin remodeling during embryogenesis [30]. In addition, MLLT3 modifies histone H3K79, which plays an important role in the development of the cerebral cortex [31]. A study on Chongming goats showed differential expression of MLLT3 between high and low fertility groups [32]. Expression of this gene is also differentially expressed in the bovine endometrium during the luteal and implantation stages, as well as before and after puberty in Bos indicus heifers [33]. The regulatory function of MLLT3 in chromatin remodeling and transcriptional elongation may provide fine-tuning of the expression of fertility-related genes, making it important for reproductive success and influencing various economically important aspects.
According to the results of our enrichment analysis, the EPHA6, ROBO1, and MLLT3 genes are involved in the following biological process library terms: GO:0007166 cell surface receptor signaling pathway, GO:0009653 anatomical structure morphogenesis, GO:0031325 positive regulation of cellular metabolic process, GO:0006464 cellular protein modification process, GO:0036211 protein modification process, GO:0009893 positive regulation of metabolic process, GO:0043412 macromolecule modification, GO:0030154 cell differentiation, GO:0048869 cellular develop-mental process, GO:0048731 system development. The obtained results, together with the data from literary sources, indicate that the EPHA6, ROBO1 and MLLT3 genes are important for the reproductive qualities of animals and can also affect other economically valuable traits. Their study in the context of genomic selection opens up new prospects for improving the fertility and productivity of farm animals.
Of particular interest is the ELAVL4 gene, which, along with EPHA6, was identified in loci under selection pressure based on the results of a comparative analysis of the genomes of Dzhalgin Merino and Australian Merino using three methods at once: FST, varLD and xp-nSL. The ELAVL4 gene encodes a protein involved in the regulation of mRNA stability and translation. The main function of ELAVL4 is to bind to mRNA in neurons and participate in post-transcriptional regulation of genes important for neuronal differentiation, development and maintenance of nervous system functions. The HuD protein, a product of the ELAVL4 gene, is a key element in the stabilization and transport of mRNA associated with neuronal development, synaptic plasticity and nervous activity [34,35]. In humans, GWAS results identify polymorphisms in the ELAVL4 gene region associated with body mass index [36]. Direct links between ELAVL4 and economically important traits in animals have not been identified at present. However, based on its role in regulating the nervous system, it is possible to assume an indirect effect on behavior and learning ability, which may be important for species with pronounced sociality or complex behavioral patterns. Also, its involvement in the formation of neuroplasticity, cognitive functions, and stress response may indirectly affect the adaptive traits of animals.
When comparing the genome of Dzhalgin Merino with the genome of Rambouillet sheep, three genes—SLC13A5, MED31 and KIAA0753—were identified at loci under selection pressure, based on the results of all three types of analysis used.
The SLC13A5 gene encodes a transport protein, the sodium-citrate cotransporter, involved in the transfer of citrate across cell membranes. Citrate plays an important role in metabolism, being a key component for the synthesis of fatty acids and maintaining energy balance, as it is involved in the tricarboxylic acid cycle (Krebs cycle) [37]. Due to the importance of the SLC13A5 gene for energy and fat metabolism, it may also be significant in the context of agricultural animal productivity, where the functioning of metabolic pathways is reflected in live weight and meat quality. The SLC13A5 gene has previously been proposed as a possible marker of intramuscular fat content in pigs [38].
The MED31 gene encodes a protein that is part of the Mediator complex, which is a multifunctional complex involved in the regulation of transcription in eukaryotes, as well as regulating key processes of cell proliferation, differentiation and organogenesis. MED31 is necessary for the correct differentiation of stem cells. When it is knocked out, there is a disruption in the differentiation of cells into certain cell lines, as has been shown in the example of adipogenesis [39]. Studies have shown that MED31 plays a key role in cell proliferation, especially at the stage of embryonic development. Thus, MED31 is involved in the regulation of the expression of genes responsible for limb development, such as Sox9 and Col2a1. These genes are necessary for chondrocyte differentiation and bone formation. Mutations in MED31 lead to disruption of the expression of these genes, which causes delays in limb rudiment development and ossification defects [40]. There is no data on the relationship between the MED31 gene and productive traits of animals; however, it can be assumed that it may be associated with the amount of fat in the carcass due to its role in the regulation of adipocyte differentiation.
The KIAA0753 gene encodes a protein involved in cell division, centriole formation, and cilia formation, which play an important role in cell polarity, signaling, and tissue differentiation. This protein is critical for the proper formation of skeletal structures, and mutations in it can lead to ciliopathies—diseases associated with disruption of cilia, which leads to multiple pathologies, including skeletal dysplasia and brain development disorders. There are no data on the association of KIAA0753 with productivity traits in animals [41,42].
It should be noted that the relatively small sample size used in this study represents a limitation that may increase the risk of bias and limit the statistical power of the analyses; therefore, the results should be interpreted with caution, although comparable sample sizes have been successfully applied in similar genomic studies of sheep and other farm animal species, particularly in research concerning local or small-sized breeds. For example, Adeniyi O.O. et al. (2022) [43], when identifying selection signatures in the Balusha Sheep Breed using the xp-EHH method, used small sample sizes: Balusha (n = 30), Bardhoka (n = 26), Istrian (n = 21), and Ruda (n = 15). As a result, genes involved in melanogenesis and T-cell receptor signaling pathways were identified, which the authors associated with selection for a specific coat color pattern and resistance to certain infectious diseases [43]. Chen S. et al. (2022) [44] detected selection signals by comparing domestic piglets of the Anqing Six-End-White breed (n = 24) with Asian Wild Boar (n = 6). Despite the small sample size, the authors were able to detect reliable selective signals, including the MSTN, SMPD4, and BCL6 genes associated with meat productivity, development, and immune response [44]. In the study by Waineina R.W. et al. (2022) [45], four goat breeds were analyzed: Galla (n = 12), Alpine (n = 29), Saanen (n = 24), and Toggenburg (n = 31). The analysis included iHS, XP-EHH, FLK, and hapFLK, which allowed the identification of loci under selection pressure containing candidate genes related to adaptation, immunity, milk production, and reproductive traits (HYAL1, HYAL3, LEPR, PDE4B, MST1, PCK) [45]. The combined use of various analytical methods to identify selection signatures with a small sample size was also applied by Dzomba et al. (2023) [46]. Their study included sheep breeds with sample sizes ranging from 8 to 52 individuals. The methods used included iHS (within-breed), XP-EHH and Rsb (between-breed), hapFLK, and FST. Despite sample sizes of less than 55 individuals, functions related to immunity, wool color, growth, and adaptation were successfully associated with the detected selection signatures [46].
A specific feature of studies on local breeds is that the available populations for analysis are often small, and forming representative samples is challenging. Nevertheless, such populations are of particular value, as their genetic structure bears traces of long-term adaptation to specific environmental conditions and the results of targeted selection. In our study, the potential limitations associated with the sample size were minimized through the application of an integrated approach combining several independent methods (FST, varLD, xp-nSL) based on different statistical principles. However, FST is a more conservative test that primarily reflects completed or ancient selection processes, accompanied by allele fixation and pronounced inter-population differentiation [47]. varLD identifies changes in local linkage disequilibrium patterns arising during population divergence and breed formation, thereby capturing selection signatures that generally reflect more recent processes than those revealed by FST [48]. In contrast, xp-nSL exhibits high power to capture recent and continuing selection processes, maintaining robustness across a wide spectrum of allele frequencies, including both low and high values [15]. These differences likely explain why the number of candidate genes identified varied among the methods. The limited overlap of the results is therefore an expected consequence of the different sensitivities of the methods and reflects distinct temporal scales of selection, whereas their combined use provides a more comprehensive and reliable identification of genomic regions under selection pressure. The integrative approach applied in this study, combining different statistical methods, enabled us to concentrate on selection signatures validated by multiple criteria. Therefore, while the limited sample size requires cautious interpretation of the findings, the applied integrated analytical approach enhances their robustness and provides valuable insights into selection signatures in the Dzhalgin Merino sheep breed.
5. Conclusions
The obtained results indicate the presence of characteristic selection signatures in the genome of the Dzhalgin Merino breed, reflecting the influence of targeted selection on the development of adaptive and productive traits. These genetic features were formed under the conditions of an arid steppe climate, which contributed to the evolution of resistance to thermal stress and adaptation to regional environmental specifics. The present study enhances our understanding of the genetic architecture underlying productive and adaptive traits in local sheep breeds and underscores the need for their further investigation and conservation as a valuable reservoir of genetic diversity and biological resilience. A promising direction for future research is the structural analysis of the identified genes located in loci under selection pressure, aimed at detecting functionally significant mutations, elucidating the mechanisms of their phenotypic effects, and identifying novel productivity markers. Thus, this study may provide a foundation for the development of new marker-assisted selection strategies for the Dzhalgin Merino breed.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abondio P. Cilli E. Luiselli D. Inferring Signatures of Positive Selection in Whole-Genome Sequencing Data: An Overview of Haplotype-Based Methods Genes 20221392610.3390/genes 1305092635627311 PMC 9141518 · doi ↗ · pubmed ↗
- 2Ma Y. Ding X. Qanbari S. Weigend S. Zhang Q. Simianer H. Properties of Different Selection Signature Statistics and a New Strategy for Combining Them Heredity 201511542643610.1038/hdy.2015.4225990878 PMC 4611237 · doi ↗ · pubmed ↗
- 3Liu Z. Bai C. Shi L. He Y. Hu M. Sun H. Peng H. Lai W. Jiao S. Zhao Z. Detection of Selection Signatures in South African Mutton Merino Sheep Using Whole-Genome Sequencing Data Anim. Genet.20225322422910.1111/age.1317335099062 · doi ↗ · pubmed ↗
- 4MészárosováM. Mészáros G. MoravčíkováN. Pavlík I. Margetín M. Kasarda R. Within-and between-Breed Selection Signatures in the Original and Improved Valachian Sheep Animals 202212134610.3390/ani 1211134635681809 PMC 9179888 · doi ↗ · pubmed ↗
- 5Eydivandi S. Roudbar M.A. Ardestani S.S. Momen M. Sahana G. A Selection Signatures Study among Middle Eastern and European Sheep Breeds J. Anim. Breed. Genet.202113857458810.1111/jbg.1253633453096 · doi ↗ · pubmed ↗
- 6Saravanan K.A. Panigrahi M. Kumar H. Bhushan B. Dutt T. Mishra B.P. Genome-Wide Analysis of Genetic Diversity and Selection Signatures in Three Indian Sheep Breeds Livest. Sci.202124310436710.1016/j.livsci.2020.104367 · doi ↗
- 7Tao L. Wang X. Zhong Y. Liu Q. Xia Q. Chen S. He X. Di R. Chu M. Combined Approaches Identify Known and Novel Genes Associated with Sheep Litter Size and Non-Seasonal Breeding Anim. Genet.20215285786710.1111/age.1313834494299 · doi ↗ · pubmed ↗
- 8Yurchenko A.A. Deniskova T.E. Yudin N.S. Dotsev A.V. Khamiruev T.N. Selionova M.I. Egorov S.V. Reyer H. Wimmers K. Brem G. High-Density Genotyping Reveals Signatures of Selection Related to Acclimation and Economically Important Traits in 15 Local Sheep Breeds from Russia BMC Genom.20192029410.1186/s 12864-019-5537-0PMC 722723232039702 · doi ↗ · pubmed ↗