Case Report: A rare RUNX1 rearrangement resulting from t(8;21)(p12;q22) in acute myeloid leukemia with plasmacytoid dendritic cell expansion
Jinying Gong, Jialong Liu, Lisha Lu, Jiru Wei, Xue Wu, Junyan Zou, Yuan Feng, Guoqing Zhu, Jing Han

TL;DR
This case report describes a rare RUNX1 gene rearrangement in a patient with a rare type of leukemia, offering insights into its possible role and treatment options.
Contribution
The paper presents the third reported case of RUNX1 rearrangement in pDC-AML, highlighting its structural and functional implications.
Findings
A RUNX1 rearrangement from t(8;21)(p12;q22) was identified in a pDC-AML patient.
The rearrangement produced a truncated RUNX1 with structural and functional similarities to RUNX1A.
CD123-targeted therapy may be a rational treatment approach for this subtype.
Abstract
In recent years, acute myeloid leukemia with plasmacytoid dendritic cell expansion (pDC-AML) has been recognized as a rare provisional subtype of AML, comprising approximately 3–5% of all reported cases and associated with a poorer clinical outcome compared with non–pDC-AML. Both RUNX1 mutations and rare rearrangements can lead to either complete loss or dominant-negative inhibition of RUNX1 function in pDC-AML, which may play a pivotal role in the aberrant expansion or malignant transformation of plasmacytoid dendritic cells (pDCs). To date, only two cases of pDC-AML with rare RUNX1 rearrangements have been reported. Herein, we reported a rare RUNX1 rearrangement resulting from t(8;21)(p12;q22) in a patient with pDC-AML, leading to the truncated RUNX1 that exhibit structural and functional similarities to RUNX1A and may act as a dominant-inhibitor of wild-type RUNX1. Given the poor…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
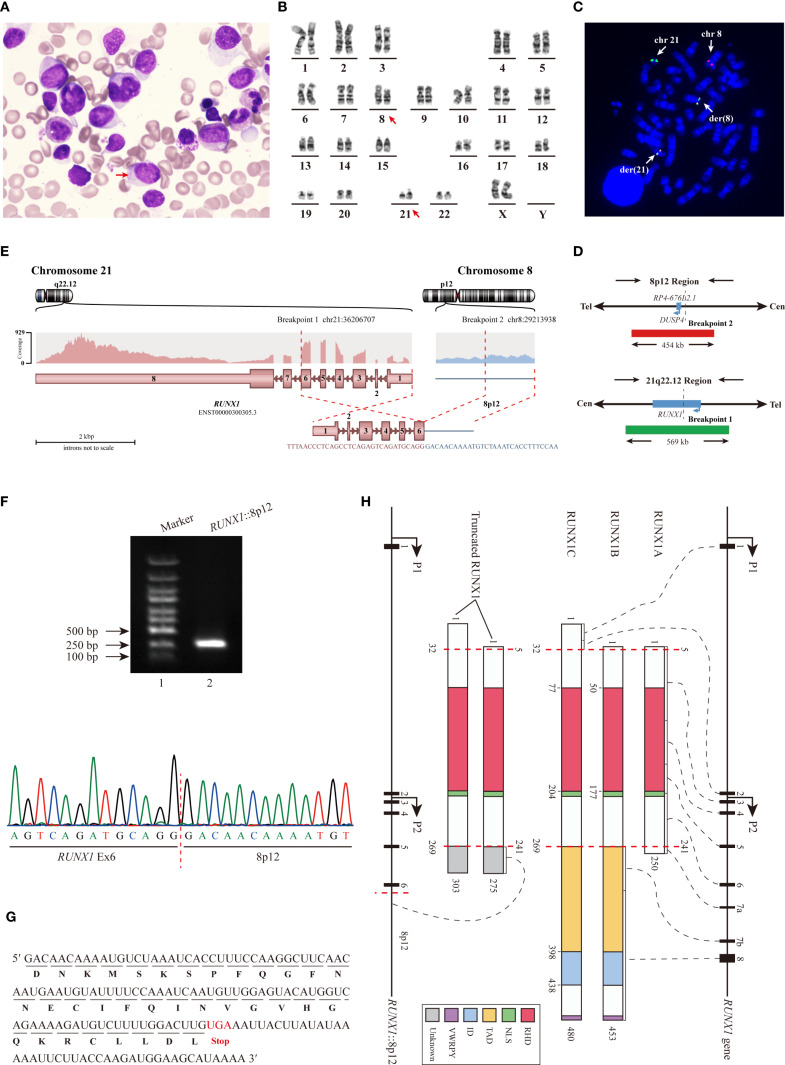 Figure 1
Figure 1| Patient | Age/sex | Partner gene | Frameshift | Reciprocal | Follow-up | Survival status | Doi |
|---|---|---|---|---|---|---|---|
| 1 | NA | (Ex8) | Out-of-frame | NA | NA | NA | 10.1073/pnas.90.16.7784 |
| 2 | 76/M | (Ex6) | Out-of-frame | No | NA | D | 10.1038/sj.leu.2403048 |
| 3 | 74/M | (Ex6) | In-frame | No | 5 m | D | 10.1002/gcc.20050 |
| 4 | 77/F | (Ex6) | Both | Yes | 2 m | R | 10.1002/gcc.20241 |
| 5 | 78/M | (Ex6) | In-frame | Yes | NA | D | 10.1182/blood-2004-07-2762 |
| 6 | 7/M | (Ex6) | In-frame | No | 10 yr | A | 10.1038/sj.leu.2404076 |
| 7 | 56/M | (Ex5/6) | In-frame | No | 2 yr | D | 10.1182/blood-2006-01-031781 |
| 8 | 73/F | (Ex5) | In-frame | NA | NA | NA | 10.1016/j.cancergencyto.2008.04.011 |
| 9 | NA | (Ex5/6) | Both | Yes | NA | NA | 10.1002/gcc.20704 |
| 10 | 1/M | (Ex3) | In-frame | No | NA | NA | 10.1002/gcc.20355 |
| 11 | 68/M | (Ex5) | In-frame | No | NA | NA | |
| 12 | 81/M | (Ex5/6) | In-frame | No | NA | NA | |
| 13 | 63/F | (Ex5/6) | In-frame | Yes | 2 yr | R | 10.1111/j.1600-0609.2007.00858.x |
| 14 | 69/M | (Ex6/7) | Out-of-frame | No | 7 m | D | 10.1038/leu.2010.106 |
| 15 | 62/M | ( | Out-of-frame | Yes | 8 m | D | |
| 16 | 63/M | (Ex6) | Out-of-frame | No | 1 m | D | 10.1016/j.cancergencyto.2010.07.116 |
| 17 | 82/F | (Ex6) | NA | NA | NA | NA | 10.1007/s12185-012-1112-z |
| 18 | 78/M | (Ex5/6) | In-frame | No | 2 yr | D | 10.1016/j.cancergen.2012.10.001 |
| 19 | 43/F | (Ex1) | Out-of-frame | No | NA | NA | 10.1002/gcc.22105 |
| 20 | 54/F | (Ex25/26) | Out-of-frame | No | 2 yr | D | 10.1186/s12943-015-0353-x |
| 21 | 76/M | (Ex5) | Out-of-frame | No | 1 yr | D | 10.1038/onc.2015.70 |
| 22 | 69/M | (Ex7) | Out-of-frame | Yes | >2 yr | A | 10.1016/j.cancergen.2017.07.002 |
| 23 | 50/M | (Ex5/6) | Out-of-frame | No | ~1 m | D | 10.1111/bjh.16444 |
| 24 | 74/F | (Ex7) | In-frame | No | 14 m | D | 10.1002/gcc.22901 |
| 25 | 80/M | (Ex2) | Out-of-frame | Yes | 5 m | D | |
| 26 | 66/M | (Ex8) | In-frame | Yes | 2 m | D | |
| 27 | 23/F | (Ex8) | Out-of-frame | No | NA | NA | |
| 28* | 61/F | (Ex2) | Out-of-frame | Yes | NA | NA | |
| 29 | 57/M | (Ex2) | In-frame | No | 19 m | D | |
| 30 | 49/M | (Ex6) | In-frame | Yes | NA | NA | |
| 31 | 33/F | (Ex2) | In-frame | No | 7 m | D | |
| 32 | 69/F | (Ex7) | In-frame | No | NA | NA | |
| 33* | 75/F | ( | NA | No | 8 m | D | |
| 34 | 1/M | (Ex5) | NA | No | NA | NA | 10.1038/s41375-023-02024-6 |
| 35 | 79/F | (Ex2) | Out-of-frame | No | 1 yr | D | 10.1007/s12308-024-00597-4 |
| 36 | 32/M | (Ex6) | In-frame | No | NA | NA | 10.1002/mc.23850 |
| 37 | 71/M | (Ex6) | NA | No | ~1 m | D | 10.1002/gcc.23272 |
| 38 | NA | (Ex5/6) | Out-of-frame | NA | NA | NA | 10.1182/blood.V97.7.2168 |
| 39 | 23/F |
| NA | NA | 1 yr | D | 10.1007/s12032-011-9890-3 |
| 40 | 7/F | (Ex7) | Out-of-frame | No | 6 m | D | 10.3892/or.2016.5119 |
| 41 | 2/M | (Ex1) | NA | Yes | 9 m | R | 10.1186/s12943-018-0881-2 |
| 42* | 51/M | (Ex6) | In-frame | No | ~2 m | D | this case |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Myeloid Leukemia Research · Chronic Lymphocytic Leukemia Research · Multiple Myeloma Research and Treatments
Introduction
In recent years, acute myeloid leukemia with plasmacytoid dendritic cell expansion (pDC-AML) has been recognized as a rare provisional subtype of AML, comprising approximately 3–5% of all reported AML cases and associated with poor clinical outcomes (1, 2). Mutations in the Runt-related transcription factor 1 (RUNX1) gene are the most frequently observed genetic alterations in pDC-AML, occurring in approximately 70% of cases, and are potentially associated with the malignant transcriptional program of plasmacytoid dendritic cells (pDCs) (1–3). Notably, RUNX1 rearrangements in pDC-AML are relatively rare, with only two cases reported to date (1, 4).
RUNX1, also known as AML1, CBFA2, or PEBP2aB, is a key transcription factor essential for the emergence of definitive hematopoiesis and the precise regulation of adult hematopoiesis, whose dysregulation can lead to aberrant hematopoietic function (5). Due to the involvement of two distinct promoters and alternative splicing in RUNX1 synthesis, the protein exists in three major isoforms: RUNX1A, RUNX1B, and RUNX1C. However, the functional relationships among these isoforms have not been fully elucidated and remain unknown (6). Recurrent genetic alterations in RUNX1 gene, primarily including monoallelic rearrangement as well as monoallelic or biallelic mutations, have been identified in both hereditary and sporadic hematologic disorders, particularly in AML or myelodysplastic syndromes (MDS) (5, 7).
RUNX1 rearrangements can give rise to two types of fusion events: gene-gene fusions, which may be either in-frame or out-of-frame, and gene-intergenic fusions (8). In-frame fusions typically generate novel fusion proteins that may act as oncogenic drivers, whereas out-of-frame and gene-intergenic fusions are more likely to result in truncated upstream gene products or potential haploinsufficiency of both genes involved (9, 10).
To date, more than 40 recurrent chromosomal rearrangements involving RUNX1 gene have been identified in AML/MDS. Among these, the most frequent partner genes are RUNX1T1 (8q21) and MECOM (3q26, including MDS1 and EVI1) (6). The RUNX1::RUNX1T1 fusion is associated with favorable prognosis, whereas the RUNX1::MECOM fusion and other rare RUNX1 fusion may be associated with unfavorable prognosis (4). Since the concept of pDC-AML was proposed around 2018 and was not recorded in the WHO classification until 2022, earlier reports of RUNX1 rearrangements may have lacked the information regarding pDCs (1, 11). This retrospective gap likely limits our current understanding of the association between RUNX1 rearrangements and pDC-AML.
Herein, we report a rare RUNX1 rearrangement resulting from t(8;21)(p12;q22) in a patient with pDC-AML, leading to the truncated RUNX1 that exhibit structural and functional similarities to RUNX1A. The truncated RUNX1 may act as a dominant inhibitor of wild-type RUNX1, potentially playing a pivotal role in the aberrant expansion or malignant transformation of pDCs. To our knowledge, this represents the third case report of RUNX1 rearrangement in pDC-AML and may provide valuable insights for future research.
Case presentation
A 51-year-old woman presented to the hospital with a six-month history of weakness and a two-week history of gingival swelling and pain. Her complete blood count indicated the following: red blood cell count of 2.96×10^12^/L, hemoglobin of 96 g/L, white blood cell count of 10.49×10^9^/L, with 38.3% monocytes and 27.9% lymphocytes, and platelet count of 181×10^9^/L. Bone marrow (BM) biopsy revealed marked proliferative activity, with a decreased proportion of granulocytes and lymphocytes, and a relative increase in monocytes. Notably, 43% of the monocytes were identified as promonocytes and immature forms, and Auer rods (red arrow) were observed in some of these cells (Figure 1A). Physical examination and computed tomography (CT) scan revealed no cutaneous lesions or lymphadenopathy, and no hepatosplenomegaly was observed.
Identification of the novel RUNX1 rearrangement. (A) BM biopsy revealed marked proliferation of promonocytes and immature monocytes. (B) Conventional chromosome analysis demonstrated the t(8;21)(p12;q22) involving the RUNX1 gene. (C) Detection of RUNX1 rearrangement by metaphase FISH Using LSI RUNX1/DUSP4 Dual Color Dual Fusion Probes. (D) Schematic diagram of LSI RUNX1/DUSP4 Dual Color Dual Fusion Probes. (E) RNA-seq identified the RUNX1::8p12 fusion. (F) RT−qPCR analysis (Lane 2) and subsequent Sanger sequencing confirmed the RUNX1::8p12 fusion. (G) Analysis of the mRNA sequence revealed an additional segment (34 aa) in the truncated RUNX1 transcript. (H) Schematic diagram illustrated the three primary RUNX1 isoforms, and the truncated RUNX1 isoforms identified in this study. Abbreviation: NLS, nuclear localization signal.
Flow cytometry analysis revealed 7.85% of abnormal myeloid blasts (CD117+, CD34+, CD13+, CD33+, CD123+, HLA-DR^dim^+ and CD38^dim^+) and 40.69% of abnormal immature monocytes (CD33^bri^+, HLA-DR^bri^+, CD38+, CD13+, CD123+, CD36+, partially CD64+, partially CD11b+, partially CD15+, partially CD14+ and CD4^dim^+). Additionally, 5.70% of pDCs were detected, with the following phenotype: CD123^bri^+, HLA-DR^bri^+, CD303+, CD304+, TDT-, CD34-, CD56-, and CD4+. The Wilm tumor gene-1 (WT1) expression was positive, with a quantitative value of 17.82%.
Conventional chromosome analysis revealed an abnormal karyotype described as 46,XX,t(8;21)(p12;q22)[20] (Figure 1B). The metaphase fluorescence in situ hybridization (FISH) analysis utilizing LSI RUNX1/RUNX1T1 Dual Color Dual Fusion Probes confirmed the presence of RUNX1 rearrangement. Part of the RUNX1 signal on chromosome 21q22 was translocated to the derivative chromosome 8. Initially, we proposed that DUSP4 gene might be the potential partner of RUNX1 gene, and the metaphase FISH utilizing LSI RUNX1/DUSP4 Dual Color Dual Fusion Probes showed a 78% positive signal (Figures 1C, D). However, RNA sequencing (RNA-seq) analysis revealed certain biases, indicating that DUSP4 was not the partner gene.
RNA-seq analysis identified the RUNX1 rearrangement event (Figure 1E). Exon 6 (Ex6) of the RUNX1 gene was fused to an intergenic region on chromosome 8p12, located approximately 5,766 base pairs upstream of the DUSP4 gene. RT−qPCR analysis and subsequent Sanger sequencing confirm the fusion between RUNX1 and the intergenic region (Figure 1F), resulting in the truncated RUNX1 fused with an additional 34 amino acid (aa) peptide of unknown function (Figure 1G). The reciprocal fusion transcript was not detected. Quantitative analysis revealed that the expression level of the fusion transcript was 32.62%.
Unfortunately, the patient was transferred to another hospital and passed away two months later, limiting the availability of further treatment information.
Discussion
In the fifth edition of the World Health Organization classification of hematolymphoid tumors (WHO-HEM5), neoplasms involving pDCs are classified into two entities: blastic plasmacytoid dendritic cell neoplasm (BPDCN) and mature plasmacytoid dendritic cell proliferation (MPDCP) associated with myeloid neoplasms (11). However, given the typical association of MPDCP with other myeloid neoplasms, the International Consensus Classification (ICC) has not formally recognized MPDCP as a distinct myeloid entity (12). Furthermore, the definition of MPDCP remains ambiguous, and the use of the term “mature” is considered inappropriate, as MPDCP cases associated with AML often comprise pDCs at early to intermediate stages of differentiation (3). The introduction of the term pDC-AML aimed to distinguish cases of MPDCP associated with AML from those involving chronic myelomonocytic leukemia (CMML) or MDS.
pDC-AML is typically characterized by cross-lineage antigen expression, adverse risk stratification, and poor outcomes, with a high frequency of RUNX1 mutations and upregulation of pDC transcriptional programs expression (1, 13). The pDCs display a spectrum of maturation from early pDCs to fully mature pDCs and are thought to originate from early pDC progenitors (14). Notably, the pDCs are predominantly at an early maturation stage. In two related studies, the expression of CD34 in pDCs from patients with pDC-AML was reported at 61% (25/41) and 98% (52/53), respectively (1, 2). In our case, pDCs were negative for CD34, suggesting that they may represent intermediate to late stages of maturation. At present, it remains unclear whether pDCs at different maturation stages have differential effects in pDC-AML.
In the study by Wenbin Xiao et al. (1), 78% (32/41) of the pDC-AML cases exhibited alterations in the RUNX1 gene, including 29 patients with RUNX1 mutations, 2 with atypical RUNX1 rearrangements, and 1 with a deletion in the region including RUNX1. Combined with data from two additional studies, the frequency of RUNX1 mutations in pDC-AML is approximately 70%, markedly higher than the 6~15% reported in overall AML (2, 3, 15) Based on the available data, the frequency of rare RUNX1 rearrangements in pDC-AML is approximately 5%, compared to less than 1% in overall AML, showing a significant increase comparable to that of RUNX1 mutations (1, 4). Therefore, we propose that rare RUNX1 rearrangements may also be associated with the development of pDC-AML.
As shown in Figure 1H, all three RUNX1 isoforms share a conserved 128 aa runt homology domain (RHD), which mediates heterodimerization with core-binding factor subunit beta (CBFB) and facilitates DNA binding to form a transcription factor complex. Additionally, RUNX1B and RUNX1C also contain a transactivation domain (TAD), an inhibitory domain (ID) and the conserved C-terminal pentapeptide motif, VWRPY. The TAD and ID regulate gene activation and repression by interacting with various proteins, while the VWRPY motif mediates transcriptional repression through interactions with Groucho/TLE transcriptional corepressors (5). RUNX1B and RUNX1C exhibit similar functions, whereas RUNX1A exerts a dominant-negative effect on both isoforms (16). Alterations in RUNX1 can generally be classified into two categories (1): those that disrupt the RHD, leading to complete loss of RUNX1 function, and (2) those that retain an intact RHD but disrupt the TAD, ID, or VWRPY motif, conferring dominant negative activity to wild-type RUNX1 (7).
Rare RUNX1 rearrangement resulting from t (8,21)(p12;q22) in our case led to the RUNX1 truncation. The predicted proteins retain the RHD but lacks the TAD, ID and VWRPY, thereby exhibiting structural and functional similarities to RUNX1A that may act as a dominant-inhibitor of wild-type RUNX1 by competing for DNA binding and interaction with CBFB (4, 16). In pDC-AML, both RUNX1 mutations (Supplementary Table 1) and rare rearrangements (Table 1) can lead to either complete loss or dominant-negative inhibition of RUNX1 function, which may play a pivotal role in the aberrant expansion or malignant transformation of pDCs. Additionally, functional suppression of RUNX1 may also occur in pDC-AML cases without detectable RUNX1 alterations.
As shown in Table 1, rare RUNX1 rearrangements, including both gene-gene and gene-intergenic fusions, are likely associated with poor prognosis. Notably, approximately half of the gene–gene fusions are out-of-frame. Since the concept of pDC-AML was proposed around 2018 and was not recorded in the WHO classification until 2022, earlier case reports may lack the information regarding pDCs (1, 11). To date, only two cases of pDC-AML with RUNX1 rearrangement have been reported (4). Compared with non–pDC-AML, pDC-AML is associated with a poorer prognosis. At present, pDC-AML remains a provisional entity within the broader category of AML, and consensus on its treatment is yet to be reached, necessitating further research.
Similar to BPDCN, pDC-AML is characterized by pDC expansion, which is associated with poor prognosis. However, the pattern of organ involvement differs between the two entities: BPDCN most commonly presents with cutaneous lesions, whereas pDC-AML predominantly affects the BM and only rarely involves the skin (1, 17). Consequently, therapeutic strategies aimed at eliminating pDCs are considered critical for both BPDCN and pDC-AML, and interleukin-3 receptor α chain (IL3RA or CD123)-targeted therapy represents a promising approach. CD123 is aberrantly overexpressed across a broad spectrum of hematologic malignancies, especially in BPDCN and AML (18). In AML, CD123 can be found in blasts, CD34+ progenitors, CD34+CD38- leukemia stem cells (LSCs), whereas normal HSCs have little (less than 1%) to no CD123 expression (18). This highly restricted expression profile makes CD123 an ideal target both for diagnostic applications and therapeutic interventions in AML, particularly in the context of pDC-AML.
Currently, tagraxofusp-erzs is the only CD123-targeted agent approved by the US Food and Drug Administration (FDA) and has demonstrated robust clinical efficacy in BPDCN, while several other CD123-targeting agents are in development or undergoing evaluation in clinical trials (18, 19). Preclinical studies in murine models have shown that tagraxofusp-erzs can effectively eliminate pDCs in pDC-AML and reduce leukemic burden, mirroring its activity in BPDCN (1). These findings suggest that CD123-directed therapy, alone or in combination with other anti-leukemic agents, may improve outcomes in pDC-AML, although its clinical efficacy has yet to be validated. Encouragingly, ongoing phase I/II clinical trials are evaluating combinations of tagraxofusp-erzs with azacitidine and venetoclax in untreated, relapsed, or refractory AML including cases of pDC-AML (NCT03113643), which may provide further insight into the therapeutic potential of CD123-targeted strategies in this disease (20). In the absence of patient transfer, we would favor this therapeutic strategy for the current case. Moreover, the t (8,21)(p12;q22) or RUNX1 rearrangement may serve as a potential marker for measurable residual disease (MRD) monitoring and assessment of therapeutic response.
In conclusion, current research on pDC-AML remains limited, and the molecular mechanisms underlying the development of pDCs are still poorly understood. Based on previous studies, signaling pathways involving the RUNX1 gene may play a critical role. Both RUNX1 mutations and rare rearrangements can result in either complete loss of RUNX1 function or exert dominant-negative effects on the wild-type RUNX1. However, the precise impact of these alterations on pDC differentiation and expansion remains unclear. It is also unknown whether certain mutations in other genes might promote pDC expansion indirectly by suppressing wild-type RUNX1. Given the poor prognosis associated with pDC-AML, further elucidation of its molecular pathogenesis is essential to guide the development of targeted therapeutic strategies and improve clinical outcomes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Xiao W Chan A Waarts MR Mishra T Liu Y Cai SF . Plasmacytoid dendritic cell expansion defines a distinct subset of RUNX 1-mutated acute myeloid leukemia. Blood. (2021) 137:1377–91. doi: 10.1182/blood.2020007897, PMID: 32871587 PMC 7955409 · doi ↗ · pubmed ↗
- 2Wang W Xu J Khoury JD Pemmaraju N Fang H Miranda RN . Immunophenotypic and molecular features of acute myeloid leukemia with plasmacytoid dendritic cell differentiation are distinct from blastic plasmacytoid dendritic cell neoplasm. Cancers (Basel). (2022) 14:3375–87. doi: 10.3390/cancers 14143375, PMID: 35884435 PMC 9324882 · doi ↗ · pubmed ↗
- 3Zalmai L Viailly PJ Biichle S Cheok M Soret L Angelot-Delettre F . Plasmacytoid dendritic cells proliferation associated with acute myeloid leukemia: phenotype profile and mutation landscape. Haematologica. (2021) 106:3056–66. doi: 10.3324/haematol.2020.253740, PMID: 33054115 PMC 8634182 · doi ↗ · pubmed ↗
- 4Aypar U Yao J Londono DM Khoobyar R Scalise A Arcila ME . Rare and novel RUNX 1 fusions in myeloid neoplasms: A single-institute experience. Genes Chromosomes Cancer. (2021) 60:100–7. doi: 10.1002/gcc.22901, PMID: 33078873 · doi ↗ · pubmed ↗
- 5Hayashi Y Harada Y Harada H . Myeloid neoplasms and clonal hematopoiesis from the RUNX 1 perspective. Leukemia. (2022) 36:1203–14. doi: 10.1038/s 41375-022-01548-7, PMID: 35354921 · doi ↗ · pubmed ↗
- 6Sood R Kamikubo Y Liu P . Role of RUNX 1 in hematological Malignancies. Blood. (2017) 129:2070–82. doi: 10.1182/blood-2016-10-687830, PMID: 28179279 PMC 5391618 · doi ↗ · pubmed ↗
- 7Bellissimo DC Speck NA . RUNX 1 mutations in inherited and sporadic leukemia. Front Cell Dev Biol. (2017) 5:111. doi: 10.3389/fcell.2017.00111, PMID: 29326930 PMC 5742424 · doi ↗ · pubmed ↗
- 8Yun JW Yang L Park HY Lee CW Cha H Shin HT . Dysregulation of cancer genes by recurrent intergenic fusions. Genome Biol. (2020) 21:166. doi: 10.1186/s 13059-020-02076-2, PMID: 32631391 PMC 7339451 · doi ↗ · pubmed ↗