Mitochondrial Dysfunction in Sickle Cell Trait Carriers With Exertional Collapse
Kristen A. Cofer, Liam Friel, Mingqiang Ren, Carolyn Dartt, Francesca Cariello, Kyung Kwon, Patricia A. Deuster, Nyamkhishig Sambuughin, Tianzheng Yu, Francis G. O'Connor

TL;DR
This study explores how mitochondrial dysfunction may contribute to exercise-induced collapse in individuals with sickle cell trait.
Contribution
The study identifies novel pathogenic mutations in mitochondrial genes linked to exertional collapse in sickle cell trait carriers.
Findings
Two Black Service Members with ECAST had pathogenic mutations in POLG and RRM2B genes.
Mitochondrial profiles in ECAST cases showed impaired function and resilience.
Mitochondrial dysfunction may play a role in exertional collapse among sickle cell trait carriers.
Abstract
Sickle cell trait (SCT) increases the risk of sudden death and exertional rhabdomyolysis (ER) in athletes and Service Members (SMs) during intense exercise. Exertional injuries in SCT carriers can result in exercise collapse associated with SCT (ECAST), an under-recognized condition characterized by variable clinical presentations ranging from ischemic muscle pain to fulminant collapse. This study presents clinical and genetic findings of two independent Black SMs with history of ECAST triggered by strenuous exercise. ECAST cases carried pathogenic heterozygous mutations POLG: p.Gly848Ser and RRM2B: p.Met282Ile associated with mitochondrial DNA depletion syndromes. Mononuclear cell mitochondria extracted from ECAST cases showed impaired mitochondrial profiles and resilience, demonstrating the potential contribution of mitochondrial dysfunction to exertional collapse in SCT carriers.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
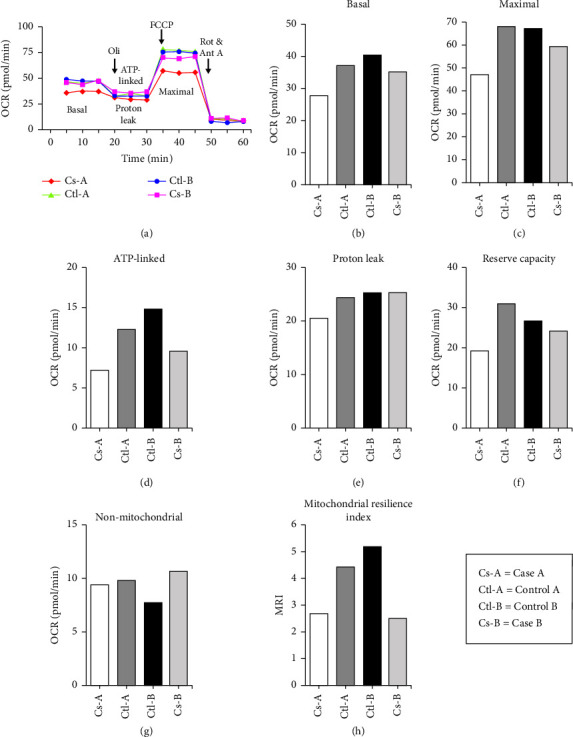 Figure 1
Figure 1- —Defense Health Agency
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHemoglobinopathies and Related Disorders · Mitochondrial Function and Pathology · Adipose Tissue and Metabolism
1. Introduction
Approximately 300 million people worldwide and nearly 7.8% of African Americans in the United States have sickle cell trait (SCT), a carrier state of mutant hemoglobin (HbAS) that in HbSS form causes sickle cell disease [1–3]. While generally considered benign, SCT has been associated with exertional injuries during intense physical exertion, especially in hot climates or high-elevation locations, or when dehydrated [1]. Exercise collapse associated with sickle cell trait (ECAST), a potentially life-threatening condition in SCT carriers, is becoming more widely recognized by the medical community. ECAST can present during or following a significant exertional effort, with unusual muscle weakness and pain, normal to modest elevation of core temperature, and an initial conscious collapse without signs of central nervous system dysfunction [4]. Although the absolute number of athletes and Service Members (SMs) who die from ECAST is extremely low, the association between sudden unexplained deaths and SCT in athletes and SMs has been increasingly recognized [4, 5]. In a study investigating the cause of sudden death cases in National Collegiate Athletic Association student athletes (2004–2008), researchers reviewed 2 million athlete years and found that African American football players with SCT had a significantly higher risk of mortality from exertion than those without SCT [5]. Before implementing safety precautions, this statistic was similar among military recruits [4, 5].
While the clinical description of ECAST is well defined, its etiology remains unclear. Existing research has focused attention on blood disorders, including alpha thalassemia and red cell pyruvate kinase deficiency [6]. This report presents two ECAST cases with pathogenic variants in nuclear genes linked to mitochondrial DNA depletion syndromes (MDS), suggesting impaired energy expenditure may contribute to exertional complications in SCT carriers.
2. Methods
This study protocol was approved by the Uniformed Services University Institutional Review Board. Four SCT-positive participants, ages 26–30 and of Black racial background, were recruited and categorized as cases (history of exertional collapse; n = 2) or controls (no history of exertional collapse; n = 2). All participants provided informed consent and completed a questionnaire detailing demographics, physical characteristics, behaviors, and medical/family histories. Cases underwent additional review of electronic medical records and a physician phone interview to gather specific exercise collapse details. Blood samples were collected from all participants for whole exome sequencing (WES) and mitochondrial profiling.
2.1. Specific Tests
2.1.1. WES
WES was performed as previously described [7]. Briefly, nonsynonymous, splice, stop gain, and stop loss variants were annotated and prioritized with minor allele frequency filtering criteria of ≤ 0.01. Variants classified as pathogenic (P), likely pathogenic (LP), variant of uncertain significance (VUS), and variants with no annotation (NA) in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) were analyzed.
2.1.2. Measurement of Mitochondrial Functions in Human Peripheral Blood Mononuclear Cells (PBMCs)
Human PBMCs were isolated from whole blood using a standard density gradient centrifugation technique, resuspended in RPMI-1640 medium. Mitochondrial oxygen consumption rate (OCR) was determined by measuring the O_2_ concentration with a Clark-type O_2_ electrode [8]. Briefly, after a basal OCR was recorded, mitochondrial proton leak was determined by adding 2-μM oligomycin (ATP synthase inhibitor). Maximal uncoupled respiration was determined by adding 2-μM carbonyl cyanide p-trifluorome-thoxyphenylhydrazone (FCCP; uncoupler), and nonmitochondrial respiration was determined by adding 1-μM rotenone (complex I inhibitor) and antimycin A (complex III inhibitor). At the end of each experiment, 5-μM rotenone was added to the PBMCs to confirm that decreases in the O_2_ concentration originated from mitochondrial respiration. A mitochondrial resilience index was calculated by dividing ATP-linked OCR and reserve capacity by proton leak and nonmitochondrial, as follows [9]:
3. Results
Demographic data and ECAST event presentation of participants are presented in Table 1.
3.1. Case A
A SM collapsed near the completion of a three-mile run. Based on presenting symptoms and peak creatine kinase, he was diagnosed with rhabdomyolysis, compartment syndrome of the left buttock, left thigh, and bilateral lower leg anterolateral compartments, and acute renal failure requiring hemodialysis. Surgical intervention included fasciotomies of the left and right lower legs, left thigh, and left buttock. WES identified heterozygous pathogenic variant p.Gly848Ser (NM_002693.3: c.G2542A, rs113994098) in the POLG gene, associated with MDS [10]. Mitochondrial profiling (Figures 1(a), 1(b), 1(c), 1(d), 1(e), 1(f), and 1(g)) showed the following OCRs in picomoles per minute (pmol/min): basal OCR = 27.67; maximal OCR = 46.67; ATP-linked OCR = 7.33; proton leak = 20.33, reserve capacity = 19; and nonmitochondrial OCR = 9.3. The mitochondrial resilience index was determined to be 2.65 (Figure 1(h)).
3.2. Case B
A SM collapsed during an indoor physical fitness test. He had a history of childhood reactive airway disease but reported no recent symptoms. WES revealed heterozygous pathogenic variant p.Met282Ile (NM_015713.5:c.846G > C, rs182614164) in the RRM2B gene, associated with MDS [10]. Mitochondrial profiling (Figures 1(a), 1(b), 1(c), 1(d), 1(e), 1(f), and 1(g)) showed the following OCRs (pmol/min): basal OCR = 35, maximal OCR = 59.33, ATP-linked OCR = 9.67, proton leak = 25.33, reserve capacity = 24.33, and nonmitochondrial OCR = 10.67. The mitochondrial resilience index was determined to be 2.49 (Figure 1(h)).
3.3. Control A
This civilian had no history of ECAST. Genetic testing for mitochondrial disorders, including MDS, was negative. Mitochondrial profiling (Figures 1(a), 1(b), 1(c), 1(d), 1(e), 1(f), and 1(g)) showed the following OCRs (pmol/min): basal OCR = 36.67; maximal OCR = 67.67; ATP-linked OCR = 12.33; proton leak = 24.33, reserve capacity = 31; and nonmitochondrial OCR = 9.67. The mitochondrial resilience index was determined to be 4.43 (Figure 1(h)).
3.4. Control B
This SM had no history of ECAST. Genetic testing for mitochondrial disorders, including MDS, was negative. Mitochondrial profiling (Figures 1(a), 1(b), 1(c), 1(d), 1(e), 1(f), and 1(g)) showed the following OCRs (pmol/min): basal OCR = 40.33; maximal OCR = 67.33; ATP-linked OCR = 15; proton leak = 25.33; reserve capacity = 27; and nonmitochondrial OCR = 7.67. The mitochondrial resilience index was determined to be 5.17 (Figure 1(h)).
4. Discussion
SCT is largely considered a benign condition. However, during intense physical exertion, in particular when accompanied by environmental heat and/or altitude, SCT carriers may be at risk for ECAST [1]. While the underlying mechanisms are not well known, current research suggests exertional sickling and/or genetic factors are contributing factors [9, 11, 12]. In this case report, we describe two SCT carriers presenting with exertional collapse events, both of whom demonstrated associated pathogenic variants linked to MDS, suggesting the potential contribution of impaired energy expenditure in ECAST.
Remarkably, two independent ECAST cases carried pathogenic mutations in the POLG and RRM2B genes. The POLG and RRM2B genes are associated with MDS [10]. MDS are genetically and clinically heterozygous autosomal recessive disorders defined by a significant reduction in mtDNA content, resulting in impaired energy production in the affected tissues and organs [10]. The POLG gene is responsible for encoding mtDNA polymerase, the enzyme that replicates the mitochondrial genome [10]. The RRM2B gene encodes ribonucleotide reductase M2 B subunit, the enzyme essential for DNA synthesis and to maintain a balanced mitochondrial nucleotide pool [10]. Mutations in both genes are also presented with a myopathic phenotype and reduced mitochondrial content in skeletal muscles [13]. While carriers of autosomal recessive mutations are typically asymptomatic, identification of two ECAST cases with similar phenotypes and carrying pathogenic mutations associated with MDS is significant. In alignment with this, the mitochondrial profiling showed that both cases exhibited impaired mitochondrial functions. Healthy mitochondria should exhibit high reserve capacity, high ATP-linked OCR, and low proton leak [9]. Conversely, our cases demonstrated low reserve capacities and low ATP-linked OCRs compared with the controls. Reduced reserve capacity may result from oxidative stress, and reduced ATP-linked OCR may indicate low ATP demand, substrate unavailability, and/or damaged oxidative phosphorylation [9]. To further differentiate between healthy and unhealthy mitochondria, the mitochondrial resilience index was calculated. Healthy mitochondria should exhibit a high mitochondrial resilience index, but both cases had lower mitochondrial resilience indices than the controls, further supporting the presence of mitochondrial dysfunctions [9].
Of note, the presence of HbS and thalassemia could represent potential sources of bias when comparing SCT carriers with and without exertional injuries. We did not measure HbS or normal hemoglobin subunit levels in this study, as our focus was on assessing mitochondrial function in PBMCs. Recent studies have shown that hemoglobin subunits can be expressed in PBMCs and may function as redox modulators [14]. Future studies are warranted to investigate the role of HbS in PBMCs and whether it significantly influences mitochondrial function. Regarding thalassemia, we performed WES on all four subjects, as described in the Methods section, and found no missense or synonymous variants in the exonic regions of the HBA1 and HBA2. All four participants carried the heterozygous HbS variant (rs344), but none carried additional exonic variants in the HBB gene.
In conclusion, we present two unrelated individuals with SCT who experienced ECAST events during exercise. Genetic analysis revealed pathogenic *POLG:*p.Gly848Ser and *RRM2B:*p.Met282Ile mutations, both associated with MDS. Mitochondrial profiling demonstrated poor mitochondrial efficiency in both cases, supporting genetic findings. While the pathophysiology of ECAST remains poorly understood, our findings suggest that genetic and mitochondrial bioenergetic analyses in individuals diagnosed with ECAST may provide valuable insight. Future research with a larger cohort should explore the role of mitochondrial dysfunctions and other genetic factors contributing to ECAST.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ren M. Sambuughin N. Mungunshukh O. Genome-Wide Analysis of Exertional Rhabdomyolysis in Sickle Cell Trait Positive African Americans Genes 2024154 p. 40810.3390/genes 1504040838674343 PMC 11049803 · doi ↗ · pubmed ↗
- 2Ojodu J. Hulihan M. M. Pope S. N. Grant A. M. Incidence of Sickle Cell Trait—United States, 2010 MMWR. Morbidity and Mortality Weekly Report 201463491155115825503918 PMC 4584538 · pubmed ↗
- 3Sambuughin N. Ren M. Capacchione J. F. Multifactorial Origin of Exertional Rhabdomyolysis, Recurrent Hematuria, and Episodic Pain in a Service Member With Sickle Cell Trait Case Reports in Genetics 201820187 November 1610.1155/2018/6898546 PMC 624765630533233 · doi ↗ · pubmed ↗
- 4O’Connor F. G. Franzos M. A. Nye N. S. Summit on Exercise Collapse Associated With Sickle Cell Trait: Finding the ‘Way Ahead’ Current Sports Medicine Reports 2021201475610.1249/jsr.000000000000080133395130 · doi ↗ · pubmed ↗
- 5Harmon K. G. Drezner J. A. Klossner D. Asif I. M. Sickle Cell Trait Associated With a RR of Death of 37 Times in National Collegiate Athletic Association Football Athletes: A Database With 2 Million Athlete-Years as the Denominator British Journal of Sports Medicine 201246532533010.1136/bjsports-2011-0908962-s 2.0-8485923902322442191 · doi ↗ · pubmed ↗
- 6Xu J. Z. Thein S. L. The Carrier State for Sickle Cell Disease Is Not Completely Harmless Haematologica 201910461106111110.3324/haematol.2018.2060602-s 2.0-8506692786131097635 PMC 6545856 · doi ↗ · pubmed ↗
- 7Sambuughin N. Mungunsukh O. Klein M. G. Genetics of Exertional Heat Illness: Revealing New Associations and Expanding Heterogeneity International Journal of Molecular Sciences 20242520 p. 1126910.3390/ijms 25201126939457051 PMC 11508780 · doi ↗ · pubmed ↗
- 8Gasier H. G. Dohl J. Suliman H. B. Piantadosi C. A. Yu T. Skeletal Muscle Mitochondrial Fragmentation and Impaired Bioenergetics From Nutrient Overload Are Prevented by Carbon Monoxide American Journal of Physiology-Cell Physiology 20203194 C 746C 75610.1152/ajpcell.00016.202032845721 · doi ↗ · pubmed ↗