Type III Congenital Pulmonary Airway Malformation: A Case Report
Maryam Soltan, Farzaneh Nayeri

TL;DR
A 4-day-old infant with a rare lung malformation required surgery after showing breathing difficulties and low oxygen levels.
Contribution
This case report adds to the clinical understanding of type III congenital pulmonary airway malformation in neonates.
Findings
The infant had a type III CPAM confirmed by surgical resection and pathological examination.
Early detection via imaging and multidisciplinary management are emphasized for neonatal CPAM.
Asymptomatic cases remain controversial in terms of treatment approach due to cancer risks.
Abstract
Congenital pulmonary airway malformation (CPAM) is a structural anomaly that occurs during development of the lower respiratory tract. We describe a 4-day-old male infant with this uncommon congenital anomaly. He presented with respiratory distress and low oxygen saturation. A chest radiograph showed infiltration in the right lower lobe, and a chest computed tomography (CT) revealed alveolar opacity with an air bronchogram pattern in the right lung along with mediastinal shift. The right lower lobe was surgically resected. Pathological examination showed an 8-cm, predominantly solid cut surface with a rare tiny cyst, consistent with a congenital cystic adenomatoid malformation (type 3). Congenital pulmonary airway malformations are the most common congenital parenchymal lung anomalies. Although their development is debated, it is believed to result from a halt in fetal bronchial tree…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
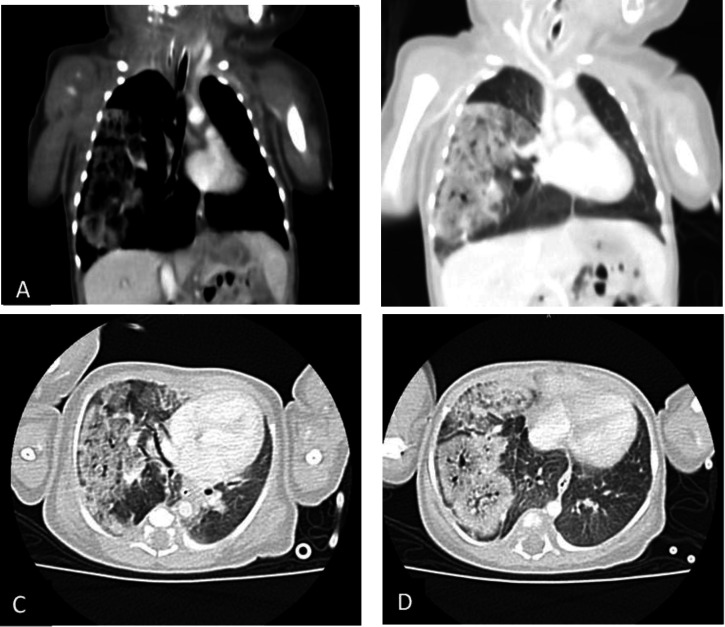 Fig. 1
Fig. 1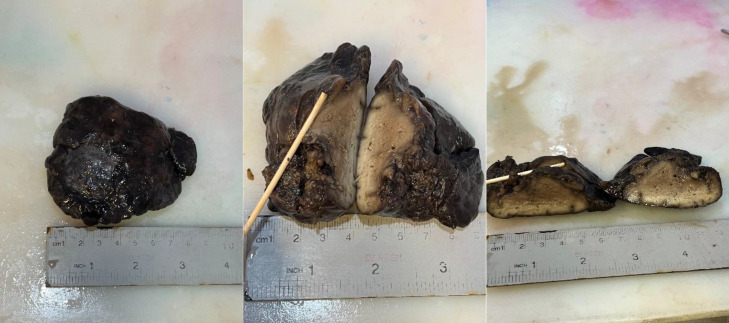 Fig. 2
Fig. 2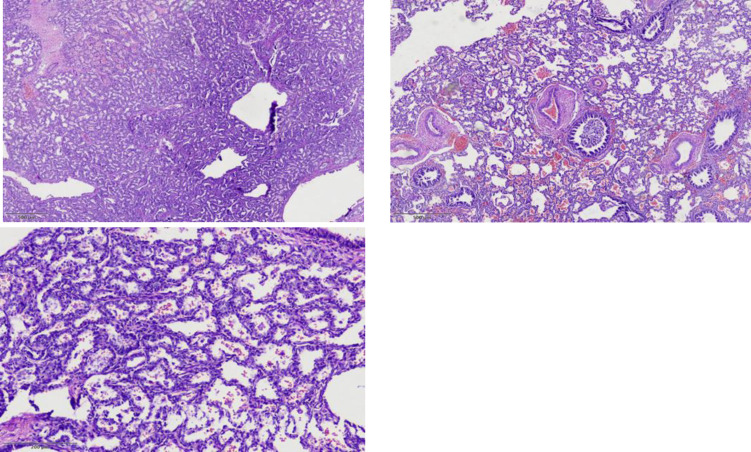 Fig. 3
Fig. 3| Type0 | Type1 | Type2 | Type3 | Type4 | |
|---|---|---|---|---|---|
| Frequency (%) | 1-3 | >65 | 20-25 | 8 | 2-4 |
| Cyst size (max cm) | 0.5 | 10 | 2.5 | 1.5 | 7 |
| Epithelial lining | Ciliated | Ciliated | Ciliated, cuboidal, or columnar | Ciliated cuboidal | Flattened alveolar lining cells |
| Muscular wall thickness (micro m) | 100-500 | 100-300 | 50-100 | 0-50 | 25-100 |
| Mucous cells | Present in all cases | Present in ~33% | Absent | Absent | Absent |
| Cartilage | Present in all cases | Present in 5-10% | Absent | Absent | Rare |
| Skeletal muscle | Absent | Absent | Present in ~5% | Absent | Absent |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCongenital Diaphragmatic Hernia Studies · Tracheal and airway disorders · Neonatal Respiratory Health Research
Introduction
Congenital parenchymal lung malformations are rare but well-characterized fetal anomalies that typically present in children. They encompass a spectrum of defects, including congenital lobar hyperinflation, bronchogenic cysts, congenital cystic adenomatoid malformation (CCAM), and lobar sequestration (1).
Here, we describe a 4-day-old male who presented with respiratory distress due to CCAM and review the literature on the clinical manifestation of this congenital lung abnormality.
Case Report
A male infant with respiratory distress and decreased oxygen saturation since birth was admitted to a referral pediatric hospital. Prenatal history and fetal ultrasound were both normal. He was delivered via cesarean section at 37 weeks of gestation, with a birth weight of 2730 g. His APGAR scores were 7 at one minute and 9 at five minutes.
Due to ongoing respiratory distress (respiratory rate of 150 breaths/min and SpO2 of 90%), a chest X-ray was performed at the birth hospital, which showed a mass (the original X-ray is not available at our facility). On admission to our hospital, arterial blood gas (ABG) analysis showed a pH of 7.42, pCO₂ of 36 mmHg, and HCO₃ of 23.4 mEq/L.
Physical examination revealed a scaphoid abdomen, and the infant was on mechanical ventilation. A chest computed tomography scan demonstrated alveolar opacity with an air bronchogram pattern in the right lung and a mediastinal shift. Diaphragmatic fluoroscopy was used to assess the integrity of the diaphragm (Figure 1).
Coronal mediastinal window(A), coronal lung window(B), and axial lung windows (C and D) reveal the near entire right lower and right middle lobes ground glass opacity with multiple low-density areas (cystic change)
Macroscopic pictures
The patient underwent a thoracotomy and right lower lobectomy at 4 days of age due to ongoing respiratory distress and concern about potential complications.
Postoperatively, the patient had a difficult recovery because of pneumonia caused by multi-resistant Klebsiella pneumoniae, leading to respiratory distress and sepsis during hospitalization. This required an extended stay in the ICU for 100 days and necessitated a thoracostomy. Ultimately, after 115 days, the patient was discharged with parental consent.
Pathological examination of the specimen revealed an 8-cm, predominantly solid congenital cystic adenomatoid malformation (CCAM), with a rare tiny cyst (Figure 2). Hematoxylin-eosin (H&E) staining showed small, irregular cystic cavities lined mainly by ciliated cuboidal to columnar epithelium. Some small cysts had thicker septa with enlarged mesenchyme and cuboidal or columnar epithelium (Figure 3).
Randomly distributed irregular bronchiole-like structures, separated by alveolus-like structures, lined by cuboidal epithelial cells imparting an adenomatoid (or gland-like) appearance.
Discussion
Congenital pulmonary airway malformations (CPAMs) encompass a range of cystic and non-cystic lung lesions that arise from early airway maldevelopment, resulting from adenomatous hyperplasia in the respiratory tract epithelium. Their incidence varies from 1:10,000 to 1:35,000 live births (1–5).
Possible differential diagnoses include congenital diaphragmatic hernia, pulmonary sequestration, bronchogenic cysts, and congenital lobar emphysema. Historically, congenital cystic adenomatoid malformations (CCAMs) were classified according to Stocker types 1, 2, and 3 (6).
“Congenital pulmonary airway malformation (CPAM)” is a more recent term that better reflects the fact that not all of these lesions are cystic (Table 1) (7). Similar to our patient, the lesion is classified as type III, which is non-cystic.
The progression of developmental defects along the tracheobronchial tree—from major airways to bronchioles and alveoli—is represented by CPAM types 0 through 4. With the increased use of ultrasound in modern obstetric practice, CPAMs are often detected during routine prenatal care; children with this condition typically present with respiratory distress and recurrent pulmonary infections (8). In our patient’s case, he presented with respiratory distress at birth and was immediately referred to a pediatric referral hospital for urgent evaluation and management.
Fetal lesions may increase or remain stable in size and can be associated with polyhydramnios or hydrops fetalis, which portend a poor prognosis (9,10). However, our patient had a normal prenatal history.
This condition may also be associated with other congenital malformations such as anasarca, renal agenesis, Potter’s syndrome, pectus excavatum, or bile duct hypoplasia, but fortunately, our patient did not exhibit any of these (11).
Surgical excision is considered the best approach to confirm diagnosis and reduce the risk of pulmonary infections and malignancy (12). There are reports of CPAM-related malignant transformations into bronchioloalveolar carcinoma, rhabdomyosarcoma, and pleuropulmonary blastoma (13). Most cases of CPAM-associated bronchioloalveolar carcinoma occur in type 1 CPAM (14).
Whether to operate on small, asymptomatic lesions should be discussed on a case-by-case basis with the parents. Despite favorable long-term outcomes in most patients, regardless of whether surgical resection is performed, long-term complications, including cancer, have been reported in patients aged 30 to 40 years (15).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Stocker JT Dail DH Hammar SP Congenital and developmental diseases Dail and Hammar's Pulmonary Pathology: Volume I: Nonneoplastic Lung Disease 2008 London Springer 13275
- 2Chong Y Rhee YJ Han SJ Cho HJ Kang SK Kang MW Life-threatening congenital cystic adenomatoid malformation in the premature neonate Korean J Thorac Cardiovasc Surg 20164932102729880210.5090/kjtcs.2016.49.3.210PMC 4900867 · doi ↗ · pubmed ↗
- 3Sfakianaki AK Copel JA Congenital cystic lesions of the lung: congenital cystic adenomatoid malformation and bronchopulmonary sequestration Rev Obstet Gynecol 2012528522866187 PMC 3410507 · pubmed ↗
- 4David M Lamas-Pinheiro R Henriques-Coelho T Prenatal and postnatal management of congenital pulmonary airway malformation Neonatology 20161102101152707035410.1159/000440894 · doi ↗ · pubmed ↗
- 5Bolde S Pudale S Pandit G Ruikar K Ingle SB Congenital pulmonary airway malformation: A report of two cases World J Clin Cases 2015354702598452310.12998/wjcc.v 3.i 5.470PMC 4419112 · doi ↗ · pubmed ↗
- 6Ribeiro FB Schultz R Congenital cystic adenomatoid malformation type I Autops Case Rep 2015532110.4322/acr.2015.019PMC 463610226558243 · doi ↗ · pubmed ↗
- 7Ursini WP Ponce CC Congenital pulmonary airway malformation Autops Case Rep 20188210.4322/acr.2018.022PMC 595318829780758 · doi ↗ · pubmed ↗
- 8Laberge JM Bratu I Flageole H The management of asymptomatic congenital lung malformations Paediatr Respir Rev 20045 Suppl AS 305121498028810.1016/s 1526-0542(04)90055-3 · doi ↗ · pubmed ↗