Transformation of Silyl‐Protected Tetrafluorinated Thia[6]helicene S‐Oxide into a Difluorinated Coronene via Induced Desilylation
Ayumu Nakao, Hiroshi Katagiri, Takashi Murase

TL;DR
A new method uses silyl protection to efficiently convert a helicene into a coronene, with insights into how silicon and fluorine influence the reaction.
Contribution
A trimethylsilyl group suppresses side reactions in thiophene S-oxide transformations, enabling efficient coronene synthesis.
Findings
TMS-protected thia[6]helicene S-oxide converts more efficiently to difluorinated coronene than unprotected forms.
Desilylation occurs via Si···F interactions and sulfur monoxide during the transformation.
Mechanistic insights reveal how silicon and fluorine control reactivity in thiophene S-oxide transformations.
Abstract
Sulfur oxidation of thiophene rings is an effective strategy for constructing substituted and polycyclic arenes via Diels–Alder reactions, wherein each thiophene ring is converted into a benzene ring. In the context of converting helicenes into planar coronenes, tetrafluorinated thia[6]helicene S,S‐dioxide undergoes a smooth intramolecular Diels–Alder reaction to afford the difluorinated coronene. However, the corresponding S‐oxide counterpart is affected by competing side reactions that limit its utility. Herein, we demonstrate that introducing a trimethylsilyl (TMS) group onto the thiophene S‐oxide ring suppresses self‐condensation and enables more efficient coronene conversion than its S,S‐dioxide counterpart. Unexpectedly, the TMS group is removed during the transformation, yielding both the TMS‐protected and deprotected forms of the difluorinated coronene. Mechanistic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
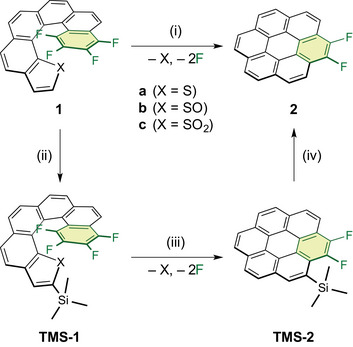 Scheme 1
Scheme 1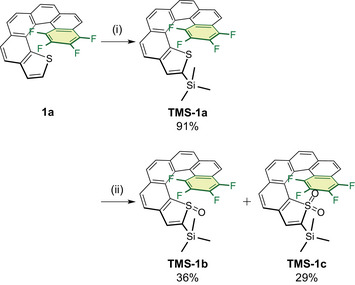 Scheme 2
Scheme 2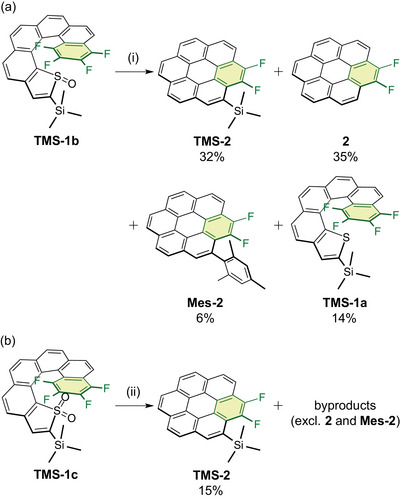 Scheme 3
Scheme 3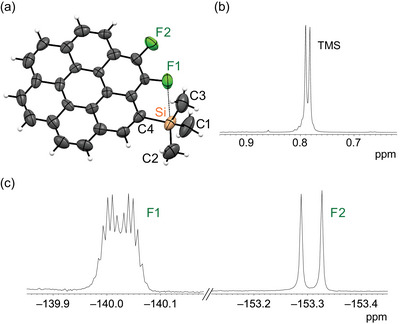 Figure 1
Figure 1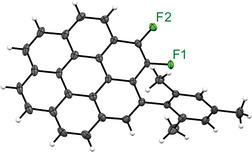 Figure 2
Figure 2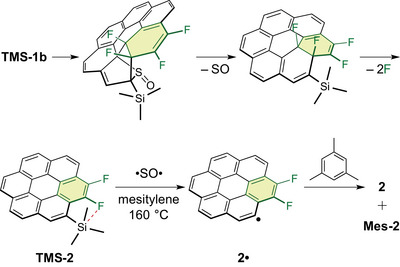 Scheme 4
Scheme 4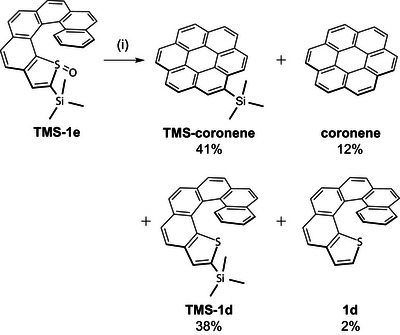 Scheme 5
Scheme 5- —The Foundation for Japanese Chemical Research
- —Network Joint Research Center for Materials and Devices (MEXT)
- —Japan Society for the Promotion of Science10.13039/501100001691
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSynthesis and Properties of Aromatic Compounds · Porphyrin and Phthalocyanine Chemistry · Photochromic and Fluorescence Chemistry
Introduction
1
Helicenes, with their screw‐shaped structures, exemplify a well‐known structural contradiction: while polycyclic aromatic systems are typically expected to be planar because of their conjugation, helicenes instead form nonplanar, fully aromatic helices.^[^ 1 ^]^ This unique geometry not only exhibits inherent chirality but also imparts helicenes with intramolecular Diels–Alder reactivity.^[^ 2, 3 ^]^ An overlooked aspect of helicene chemistry is their ability to transform into planar structures under specific external stimuli, even without the addition of reagents. For example, [5]helicene can be converted into benzo[ghi]perylene through photochemical cyclodehydrogenation,^[^ 4 ^]^ or even thermal cyclodehydrogenation using flash vacuum pyrolysis.^[^ 5 ^]^ Remarkably, heating [6]helicene to 485 °C in a sealed tube induces its planarization, yielding trace amounts of coronene.^[^ 6 ^]^ These transformations illustrate the structural versatility of helicenes, bridging the gap between nonplanar and planar polycyclic aromatic systems. However, their limited efficiency in the case of [6]helicene highlights the challenges that remain in realizing their synthetic potential.
To address the inefficient transformation of [6]helicene into coronene, we recently demonstrated that replacing one of its terminal benzene rings with a thiophene ring having the sulfur atom oriented inwards enabled its photochemical transformation into coronene.^[^ 7 ^]^ The introduction of four fluorine atoms to the opposite terminal benzene ring produced F_4_‐thia[6]helicene 1a, which underwent facile photochemical transformation into F_2_‐coronene 2 (Scheme 1, route (i)). The corresponding thermal transformation is facilitated by S‐oxidation of the thiophene ring to its S,S‐dioxide form. Specifically, heating F_4_‐thia[6]helicene S,S‐dioxide 1c in mesitylene at 160 °C afforded 2 in 55% yield,^[^ 7 ^]^ representing a significant improvement in efficiency compared with the reaction of the parent [6]helicene. Although the S‐oxide form 1b also undergoes this “helix‐to‐disc” conversion, its efficiency is reduced by the effect of competing side reactions, including deoxygenation of the thiophene S‐oxide ring and self‐condensation of 1b via a Diels–Alder reaction.
“Helix‐to‐disc” conversion of F4‐thia[6]helicene 1 into F2‐coronene 2. (i, iii) A cascade reaction triggered by an intramolecular Diels–Alder reaction, followed by cheletropic extrusion of sulfur‐containing species and elimination of two fluorine atoms, (ii) silylation of the thiophene ring, and (iv) desilylation at the coronene framework. Helicene structures are depicted in the M configuration for clarity; however, all samples were used as racemic mixtures.
As revealed by our prior DFT analysis,^[^ 7 ^]^ the transformation of 1c into 2 begins with an intramolecular Diels–Alder reaction. The calculations showed that the LUMO is found on the thiophene S,S‐dioxide ring (diene) and the HOMO on the F_4_‐benzene ring (dienophile), suggesting an inverse electron‐demand nature. Favorable overlap of these frontier orbitals, enforced by the helicene topology, dictates the regioselectivity of the [4+2] cycloaddition. This cycloaddition is followed by spontaneous cheletropic extrusion of sulfur dioxide (SO_2_) and elimination of two fluorine atoms. A similar sequence is proposed for the transformation of 1b into 2, except for the extrusion of sulfur monoxide (SO). Although susceptible to side reactions, 1b was calculated to have a lower activation free energy for the intramolecular Diels–Alder reaction, indicating higher intrinsic reactivity than 1c.
Given that a thiophene S‐oxide ring is generally considered more favorable for Diels–Alder reactions than a thiophene S,S‐dioxide ring,^[^ 8 ^]^ the reduced efficiency of 1b in its transformation into 2 was unexpected. Indeed, thiophene S‐oxides are known to act as useful dienes, affording substituted arenes.^[^ 9 ^]^ A striking example of this reactivity is the synthesis of hexaarylbenzenes with six different substituents.^[^ 10 ^]^ This transformation was achieved through Diels–Alder reactions between tetraarylthiophene S‐oxides and diarylethynes, enabling regioselective aryl group installation on the central benzene core. Notably, the corresponding tetraarylthiophene S,S‐dioxides did not undergo Diels–Alder reactions with diarylethynes. This result highlights the distinct reactivity of substituted thiophene S‐oxides, which are inherently nonplanar in conformation.^[^ 11 ^]^ Thus, substituted thiophene S‐oxides retain their high reactivity while suppressing self‐condensation, enabling the desired transformations.
Herein, we report trimethylsilyl (TMS)‐protected F_4_‐thia[6]helicene S‐oxide TMS‐1b, which bears a TMS group at the terminal thiophene S‐oxide ring (Scheme 1, route (ii)). Among silyl protecting groups, TMS was selected for its minimal steric hindrance during coronene conversion and facile removal afterward (Scheme 1, routes (iii, iv)). TMS‐1b exhibited higher efficiency in coronene conversion than 1c, despite the steric hindrance introduced by the TMS group, which is absent in 1c. Furthermore, no self‐condensation was observed. Unexpectedly, TMS cleavage occurred during the transformation into TMS‐2, yielding not only TMS‐2 but also 2. X‐ray crystallographic analysis of TMS‐2 provided insight into this desilylation process. The close proximity of the Si and F atoms in TMS‐2 highlights a key structural feature of this transformation, shedding light on their roles in the reaction mechanism.
Results and Discussion
2
Synthesis of TMS‐1
2.1
Thiahelicenes have been synthesized via various methods, among which the pioneering work by Wynberg et al. demonstrated the effectiveness of stilbene‐type photocyclization.^[^ 12 ^]^ This approach has since been widely utilized, predominantly yielding thiahelicenes with sulfur atoms oriented outwards, with octathia[15]helicene, the longest known thiahelicene, being a representative case.^[^ 13 ^]^ However, inward‐oriented thiahelicenes have been less explored, although recent studies by Guijarro et al. have demonstrated their synthesis via photocyclization,^[^ 14 ^]^ as exemplified by the preparation of 1a in this study.
One advantage of thiahelicenes is their ability to undergo regioselective functionalization at the terminal thiophene ring.^[^ 15 ^]^ Such functionalization can be achieved through direct electrophilic substitution or via the generation of α‐anions followed by reaction with electrophiles, enabling precise and versatile modifications. The functionalization of 1a was carried out using lithium diisopropylamide (LDA), followed by the addition of TMS‐Cl, to afford TMS‐1a in 91% yield (Scheme 2). The bulky LDA base was selected instead of the conventional n‐BuLi to avoid nucleophilic aromatic substitution at the terminal F_4_‐benzene ring.^[^ 16 ^]^
Synthesis of TMS‐1. Conditions: (i) LDA, TMS‐Cl, THF, −80 °C, 4 h; (ii) m‐CPBA (1.0 equiv.), CH2Cl2, 0 °C, 5 h.
The selective oxidation of a thiophene ring to its S‐oxide is typically achieved using m‐CPBA in the presence of BF_3_·Et_2_O.^[^ 17 ^]^ Using this method, 1b was prepared from 1a in 97% yield.^[^ 7 ^]^ However, applying this method to TMS‐1a resulted in the rapid removal of the TMS group, regenerating 1a. Therefore, TMS‐1a was treated with m‐CPBA (1.0 equiv.) in the absence of BF_3_·Et_2_O, affording a mixture of TMS‐1b (36%), TMS‐1c (29%), and unreacted TMS‐1a (29%). The TMS‐functionalized products were readily separated by column chromatography on silica gel with CHCl_3_, eluting in the following order: TMS‐1a < TMS‐1c < TMS‐1b. Notably, TMS‐1b exhibited enhanced stability in solution, resisting self‐condensation and deoxygenation for over one month at room temperature (Figure S1). In contrast, 1b gradually underwent these side reactions under the same conditions, demonstrating the stabilizing effect of the TMS group.
Conversion Into TMS‐2
2.2
With the thermally stable TMS‐1b in hand, we next performed its coronene conversion in mesitylene at 160 °C (Scheme 3a). After heating for 30 min, the starting material was completely consumed, yielding TMS‐2 (32%), 2 (35%), and Mes‐2 (6%), in which the TMS group was removed and solvent mesitylene was attached onto the coronene core. Additionally, deoxygenation of the thiophene S‐oxide ring afforded TMS‐1a (14%). Hence, the combined yields of TMS‐2, 2, and Mes‐2 resulted in an overall coronene conversion of 73%, surpassing the 55% yield obtained from 1c. In contrast, TMS‐1c required prolonged heating (80 min) for complete consumption, yet the coronene conversion yield was significantly lower (15%), accompanied by numerous complex byproducts, excluding 2 and Mes‐2 (Scheme 3b). These results indicate that the TMS group retarded coronene conversion but preserved the intrinsic high reactivity of the thiophene S‐oxide ring. Unlike 2, which adopts a completely planar structure, TMS‐2 and Mes‐2 exhibit enhanced solubility in CH_2_Cl_2_ and CHCl_3_ owing to the steric effects of the TMS and mesityl groups.
(a,b) Thermal transformation of TMS‐1b and TMS‐1c into TMS‐2. Conditions: (i) mesitylene, 160 °C, 30 min; (ii) mesitylene, 160 °C, 80 min.
The structure of TMS‐2 was unambiguously determined by X‐ray crystallographic analysis (Figure 1a).^[^ 18 ^]^ The Si atom was in close contact with the F1 atom (Si···F1, 2.842 Å), forming a pseudo‐pentacoordinate Si center.^[^ 19 ^]^ This distance was considerably shorter than the sum of the van der Waals radii of silicon and fluorine (3.57 Å), suggesting a significant Si···F interaction. Notably, the C2–Si bond (1.903 Å), which was positioned trans to the F1 atom, was longer than the C1–Si bond (1.843 Å) and the C3–Si bond (1.840 Å) in the TMS group. Similarly, the aromatic C4–Si bond (1.895 Å) was elongated and weakened, resembling the C2–Si bond. The close proximity of the F1 atom influenced the NMR spectra of TMS‐2 (Figure 1b, c). The ^1^H NMR signal of the TMS protons appeared as a doublet at δ = 0.79 ppm because of long‐range H–F1 coupling (^6^ J HF = 4.1 Hz). The ^19^F NMR signal of the F1 atom was distinctive, appearing as a doublet of decets, with an initial doublet splitting because of vicinal F1–F2 coupling (^3^ J FF = 18.2 Hz), followed by further splitting into a decet caused by the nine protons of the TMS group.
(a) Crystal structure of TMS‐2. Thermal displacement ellipsoid plot of TMS‐2 at the 50% probability level. Selected interatomic distances (Å): C1–Si, 1.843(9); C2–Si, 1.903(8); C3–Si, 1.840(8); C4–Si, 1.895(5); Si···F1, 2.842(4). (b) 1H NMR spectrum (500 MHz, 298 K, CDCl3) of TMS‐2 (TMS region only). (c) 19F NMR spectrum (470 MHz, 298 K, CDCl3) of TMS‐2.
The X‐ray crystal structure of Mes‐2 provided conclusive evidence that solvent mesitylene had indeed bound to the coronene core, confirming the unexpected formation of the arylated product (Figure 2).^[^ 18 ^]^ In the ^1^H NMR spectrum of Mes‐2, the aromatic protons of the mesityl group appeared at 7.15 ppm and its methyl protons resonated at 2.50 and 2.04 ppm with an intensity ratio of 1:2. Unlike the TMS methyl protons of TMS‐2, these singlet signals indicated that the mesityl methyl protons were distant from the fluorine atoms and that the mesityl group underwent restricted rotation around the coronene core on the NMR timescale. In the ^19^F NMR spectrum, the F1 and F2 atoms appeared at −146.5 and −152.2 ppm, respectively, behaving as doublets because only vicinal F1–F2 coupling occurred. This simple splitting pattern for the F1 atom contrasted with the more complex signal observed for F1 in TMS‐2 (Figure 1c).
Crystal structure of Mes‐2. Thermal displacement ellipsoid plot of Mes‐2 at the 50% probability level.
First, we hypothesized that the removal of the TMS group during the transformation of TMS‐1b into TMS‐2 at 160 °C occurred spontaneously because of the weakened aromatic C4–Si bond in TMS‐2. However, when isolated, TMS‐2 remained intact even after heating at 160 °C for 2 h, suggesting that thermal cleavage of the C4–Si bond alone was insufficient to account for the TMS group removal. Given that two fluorine atoms were eliminated during the transformation, we next hypothesized that the liberated fluorine species interacted with the Si atom, facilitating the cleavage of the C4–Si bond. Indeed, treatment of TMS‐2 with Bu_4_NF in THF at room temperature effected facile desilylation within 3 h, affording 2 in quantitative yield (Scheme 1, route (iv)). However, during the transformation of TMS‐1c into TMS‐2, no desilylation was observed, even though the same elimination of two fluorine atoms occurred (Scheme 3b). Therefore, the liberated fluorine species were not fluoride anions and did not contribute to the cleavage of the C4–Si bond.
As with 1b and 1c,^[^ 7 ^]^ the transformation of TMS‐1b and TMS‐1c into TMS‐2 is also expected to be initiated by the inverse electron‐demand intramolecular Diels–Alder reaction between the TMS‐protected S‐oxidized thiophene ring (diene) and the F_4_‐benzene ring (dienophile) (Scheme 4). The key difference between the two systems lies in the subsequent extrusion step: the former releases highly reactive SO, whereas the latter releases thermodynamically stable SO_2_. In both cases, these extrusion processes are accompanied by the elimination of two bridgehead fluorine atoms, driven by aromatization toward the coronene framework. The formation of Mes‐2 as a byproduct suggests the involvement of radical intermediates, since such reactivity cannot be readily explained by ionic or concerted mechanisms. We therefore propose that SO, which exhibits diradical character similar to O_2_,^[^ 20 ^]^ induces homolytic cleavage of the aromatic C–Si bond in TMS‐2 under the present reaction conditions (160 °C, mesitylene) (Scheme 4). Upon TMS removal, the resulting F_2_‐coronenyl radical 2• can either abstract a hydrogen atom from the solvent mesitylene to yield 2 or undergo direct arylation with mesitylene to form Mes‐2. The radical nature of SO, particularly its reactivity at the S atom, is reminiscent of thiyl radicals (RS•), which have recently been reported to mediate radical desilylation of arylsilanes.^[^ 21 ^]^ This resemblance further supports the involvement of SO in TMS removal via a radical pathway.
Proposed mechanism for the formation of 2 and Mes‐2 from TMS‐1b via homolytic cleavage of the aromatic C–Si bond in TMS‐2, assisted by Si···F interactions. •SO• is the notation used to emphasize that SO exhibits diradical character. Note that TMS‐2 remains stable at 160 °C in mesitylene under conditions where SO is not generated.
Impact of Fluorine on the TMS Group Removal
2.3
Deoxygenated TMS‐1a was detected in the reaction mixture of TMS‐1b, but no further desilylation to 1a was observed (Scheme 3a), suggesting that Si···F interactions play a significant role in promoting TMS group removal. To further investigate the role of fluorine, we examined the coronene conversion of the non‐fluorinated S‐oxide TMS‐1e (Scheme 5). Heating TMS‐1e in mesitylene at 160 °C for 3 h afforded TMS‐coronene (41%) and coronene (12%), corresponding to 53% coronene conversion, which is lower than the 73% observed for TMS‐1b. Desilylation also occurred in the absence of fluorine, confirming that the fluorine atoms eliminated during the transformation of TMS‐1b were not directly responsible for TMS cleavage. Instead, desilylation is likely facilitated by in‐situ‐generated SO. Prolonged heating led to unavoidable deoxygenation of the thiophene S‐oxide ring, yielding TMS‐1d (38%) and its desilylated counterpart 1d (2%).
Thermal transformation of TMS‐1e. Conditions: (i) mesitylene, 160 °C, 3 h.
The desilylation behavior between the fluorinated and non‐fluorinated systems showed a marked contrast. In the fluorinated system, desilylation occurred selectively at TMS‐2, whereas in the non‐fluorinated system, it proceeded from both TMS‐coronene and TMS‐1d. Furthermore, the extent of desilylation was significantly lower in the non‐fluorinated system: the total yield of desilylated products was 14% (coronene: 12%, 1d: 2%), whereas it reached 41% in the fluorinated system (2: 35%, Mes‐2: 6%). Since SO is expected to be generated in both systems, this contrast indicates that SO alone cannot account for the observed desilylation behavior. As shown in Scheme 4, Si···F interactions are proposed to act synergistically with SO to promote TMS removal. As previously observed in the TMS‐free system,^[^ 7 ^]^ fluorine substitution also accelerated coronene conversion, with TMS‐1e requiring 3 h for completion versus just 30 min for TMS‐1b. These experimental results collectively demonstrate that fluorine plays a dual role: enhancing the efficiency of the coronene transformation and assisting SO‐induced desilylation through Si···F interactions.
Conclusion
3
In summary, we demonstrated that TMS‐protected F_4_‐thia[6]helicene S‐oxide TMS‐1b undergoes a highly efficient “helix‐to‐disc” conversion under thermal conditions. While thiophene S‐oxides are inherently more reactive than their S,S‐dioxide counterparts, the transformation of F_4_‐thia[6]helicene S‐oxide 1b into F_2_‐coronene 2 had previously been challenging because of competing side reactions. In this study, the introduction of a TMS group at the thiophene S‐oxide ring was found to prevent its self‐condensation, thereby enabling its selective coronene conversion. Note that the TMS group was eliminated during the transformation, yielding not only TMS‐2 but also 2 and Mes‐2, the latter featuring a mesityl group attached to the coronene core from solvent mesitylene. Mechanistic investigations indicated that this unexpected desilylation results from the combined effect of Si···F interactions and in‐situ‐generated SO. The suppressed desilylation in the non‐fluorinated system highlights the crucial role of fluorine in promoting TMS group removal. These findings provide new insights into the distinct roles of silicon, fluorine, and in‐situ‐generated SO in facilitating cascade transformations of helicenes and expand the synthetic utility of thiophene S‐oxides in the development of functionalized polycyclic aromatic systems.
Experimental Section
4
Thermal transformation of TMS‐1b
A mesitylene solution of TMS‐1b (27.8 mg, 0.0562 mmol in 28 mL of mesitylene, 2.0 mM) was heated at 160 °C for 30 min. After confirming completion of the reaction by TLC, the solution was passed through a silica gel column (hexane/toluene = 10:1), then triturated with MeOH (2 × 1 mL) and hexane/CHCl_3_ (3:1, 2 × 4 mL). The resulting precipitate was collected and further purified by recycling preparative GPC (CHCl_3_) to give TMS‐2 (7.31 mg, 0.0179 mmol, 32%), 2 (6.62 mg, 0.0197 mmol, 35%), Mes‐2 (1.50 mg, 3.30 × 10^−3^ mmol, 6%), and TMS‐1a (3.88 mg, 8.11 × 10^−3^ mmol, 14%).
TMS‐2: pale yellow solid; TLC (hexane/CHCl_3_ = 3:1) R f = 0.68; M.p. 268.5–270.0 °C; ^1^H NMR (500 MHz, CDCl_3_, 298 K) δ = 9.11 (s, 1H), 8.81 (d, 1H, J = 8.6 Hz), 8.73 (d, 1H, J = 8.4 Hz), 8.70 (d, 1H, J = 8.4 Hz), 8.66 (d, 1H, J = 8.4 Hz), 8.64 (d, 1H, J = 8.4 Hz), 8.63 (d, 1H, J = 8.6 Hz), 8.61 (d, 1H, J = 8.4 Hz), 8.56 (d, 1H, J = 8.4 Hz), 0.79 (d, 9H, ^6^ J HF = 4.1 Hz); Chemical shifts highly depend on the sample concentration; ^19^F NMR (470 MHz, CDCl_3_, 298 K) δ = −140.0 (doublet of decets, 1F, ^3^ J FF = 18.2 Hz, ^6^ J HF = 4.1 Hz), −153.3 (d, 1F, ^3^ J FF = 18.2 Hz); ^13^C{^1^H, ^19^F} NMR (125 MHz, CDCl_3_, 298 K) δ = 145.1 (CF), 144.3 (CF), 135.6 (CH), 131.9 (C), 129.1 (C), 128.5 (C), 128.0 (C), 127.3 (C), 126.8 (CH), 126.4 (CH), 126.3 (CH+CH+CH), 126.00 (CH), 125.96 (CH), 123.0 (C), 122.0 (C), 121.6 (C), 121.3 (C+C), 119.6 (C), 119.4 (C), 119.2 (C), 117.7 (CH), 1.14 (SiMe_3_); HR‐MS (APCI) m/z calcd for [M]^+^: 408.1140, found: 408.1135.
Mes‐2: pale orange solid; TLC (hexane/CHCl_3_ = 3:1) R f = 0.63; M.p. > 285.0 °C (decomp.); ^1^H NMR (500 MHz, CDCl_3_, 298 K) δ = 9.09 (d, 1H, J = 8.7 Hz), 8.99 (d, 1H, J = 8.8 Hz), 8.97 (d, 1H, J = 8.5 Hz), 8.96 (d, 1H, J = 8.4 Hz), 8.94 (d, 1H, J = 8.1 Hz), 8.94 (d, 1H, J = 8.1 Hz), 8.92 (d, 1H, J = 8.5 Hz), 8.89 (d, 1H, J = 8.5 Hz), 8.72 (s, 1H), 7.15 (s, 2H), 2.50 (s, 3H), 2.04 (s, 6H); Chemical shifts highly depend on the sample concentration; ^19^F NMR (470 MHz, CDCl_3_, 298 K) δ = −146.5 (d, 1F, ^3^ J FF = 17.6 Hz), −152.2 (d, 1F, ^3^ J FF = 17.6 Hz); ^13^C{^1^H, ^19^F} NMR (125 MHz, CDCl_3_, 298 K) δ = 145.0 (CF), 144.6 (CF), 139.2 (C), 136.9 (C), 136.1 (2C), 134.0 (C), 129.4 (CH), 129.2 (C), 129.0 (C), 128.6 (C), 128.5 (C), 127.9 (2CH), 127.4 (CH), 126.9 (CH), 126.8 (CH), 126.6 (CH), 126.52 (CH), 126.50 (CH), 126.2 (CH), 122.2 (C), 122.1 (C), 122.0 (C), 121.5 (C), 120.8 (C), 120.0 (C), 119.9 (C), 118.9 (C), 118.2 (CH), 21.3 (CH_3_), 20.9 (2CH_3_); HR‐MS (APCI) m/z calcd for [M+H]^+^: 455.1606, found: 455.1602.
Thermal transformation of TMS‐1c
A mesitylene solution of TMS‐1c (28.1 mg, 0.0550 mmol in 27.5 mL of mesitylene, 2.0 mM) was heated at 160 °C for 80 min. After confirming completion of the reaction by TLC, the reaction mixture was concentrated in vacuo. The resulting residue was passed through a silica gel column (hexane/toluene = 10:1), then triturated with MeOH (2 × 1 mL). The precipitate was collected and further purified by recycling preparative GPC (CHCl_3_) to give TMS‐2 (3.3 mg, 8.1 × 10^−3^ mmol, 15%). The reaction also gave numerous complex byproducts, but thorough analysis confirmed that neither 2 nor Mes‐2 was present.
TMS‐Deprotection of TMS‐2
TMS‐2 (5.0 mg, 0.012 mmol) was dissolved in THF (0.25 mL), and Bu_4_NF in THF (1 M, 0.040 mL) was added dropwise. The resulting solution was stirred at room temperature for 3 h. The reaction mixture was then quenched with saturated NH_4_Cl solution (3 mL), and the resulting precipitate was triturated with water (2 × 3 mL) to give 2 (3.3 mg). The supernatant was extracted with toluene, and the combined organic layer was concentrated. The resulting residue was further triturated with water (2 × 1 mL) to give an additional portion of 2 (0.7 mg). In total, 2 was obtained quantitatively as a pale yellow solid (4.0 mg, 0.012 mmol), showing analytical purity in its NMR spectra.
Thermal transformation of TMS‐1e
A mesitylene solution of TMS‐1e (19.9 mg, 0.0471 mmol in 23.5 mL of mesitylene, 2.0 mM) was heated at 160 °C for 3 h. After confirming completion of the reaction by TLC, the solution was passed through a silica gel column (hexane) and triturated with MeOH (2 × 1 mL). The resulting precipitate was collected and found to contain TMS‐coronene and coronene. The supernatant was concentrated in vacuo, and the residue was purified by column chromatography on silica gel (hexane) to give TMS‐coronene, coronene, TMS‐1d, and 1d. In total, TMS‐coronene (7.14 mg, 0.0192 mmol, 41%), coronene (1.72 mg, 5.73 × 10^−3^ mmol, 12%), TMS‐1d (7.33 mg, 0.0180 mmol, 38%), and 1d (0.33 mg, 9.87 × 10^−4^ mmol, 2%) were obtained.
TMS‐coronene: pale yellow solid; TLC (hexane/CHCl_3_ = 3:1) R f = 0.61; M.p. 256.0–258.0 °C; ^1^H NMR (500 MHz, CDCl_3_, 298 K) δ = 9.18 (d, 1H, J = 8.7 Hz), 9.11 (s, 1H), 8.93 (d, 1H, J = 8.7 Hz), 8.90–8.85 (m, 8H), 0.82 (s, 9H); Chemical shifts highly depend on the sample concentration; ^13^C{^1^H} NMR (125 MHz, CDCl_3_, 298 K) δ = 136.5 (C), 133.6 (CH), 132.3 (C), 129.0 (C), 128.7 (C), 128.6 (C), 128.3 (C), 127.8 (C), 126.6 (CH), 126.34 (CH), 126.27 (CH), 126.19 (CH), 126.15 (CH+CH), 126.2 (CH), 126.1 (CH), 126.0 (CH), 125.7 (CH), 123.2 (C), 123.0 (C), 122.7 (C), 122.53 (C), 122.45 (C), 122.4 (C), 0.72 (SiMe_3_); HR‐MS (APCI) m/z calcd for [M]^+^: 372.1329, found: 372.1332.
Supporting Information
The authors have cited additional references within the Supporting Information.^[^ 22, 23, 24, 25 ^]^
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Supporting Information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1a) Y. Shen , C.‐F. Chen , Chem. Rev. 2012, 112, 1463;22017405 10.1021/cr 200087 r · doi ↗ · pubmed ↗
- 2a) R. H. Martin , J. Jespers , N. Defay , Helv. Chim. Acta 1975, 58, 776;
- 3a) T. Murase , C. Matsuda , K. Adachi , T. Sawada , M. Fujita , Commun. Chem. 2018, 1, 97;
- 4a) F. B. Mallory , C. W. Mallory , in Organic Reactions, Vol. 30 (Ed.: W. G. Dauben ), Wiley‐VCH, Weinheim, 1984, p. 51;
- 5X. Xue , L. T. Scott , Org. Lett. 2007, 9, 3937.17760450 10.1021/ol 7015516 · doi ↗ · pubmed ↗
- 6R. C. Dougherty , J. Am. Chem. Soc. 1968, 90, 5788.
- 7K. Seino , T. Okano , K. Oya , H. Katagiri , T. Murase , Chem. Eur. J. 2024, 30, e 202402445.39051923 10.1002/chem.202402445 · doi ↗ · pubmed ↗
- 8a) A. S. Cieplak , J. Am. Chem. Soc. 1981, 103, 4540;