The emerging view on the roles of butyrate in Clostridioides difficile pathogenesis
Horia A. Dobrila, Andrew J. Hryckowian

TL;DR
This paper explores how butyrate, a gut microbiome metabolite, influences Clostridioides difficile infections and could lead to new treatment strategies.
Contribution
The paper reviews emerging evidence on butyrate's role in C. difficile pathogenesis and identifies gaps in current knowledge.
Findings
Butyrate-rich gut environments may exclude Clostridioides difficile.
Butyrate impacts C. difficile growth, metabolism, toxin production, and sporulation.
Understanding butyrate's role could lead to new strategies for mitigating CDI.
Abstract
The Centers for Disease Control and Prevention classifies Clostridioides difficile as an urgent threat to the nation’s health, as it causes 450,000 infections, 15,000 deaths, and 1 billion dollars in excess healthcare costs per year in the United States. Current treatments for C. difficile infections (CDIs) are antibiotics and, in recurrent cases, microbiome restoration therapy (MRT). Antibiotics contribute to antibiotic resistance and recurrent CDIs. Although MRTs (e.g., defined consortia of microbes or fecal transplant) are increasingly accessible, the long-term sustainability and accessibility of these treatments remain to be determined. These limitations highlight the need for more precise strategies for coping with CDI. Because a disrupted (dysbiotic) gut microbiome is the primary risk factor for CDI, a better understanding of the interactions between C. difficile, the microbiome,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
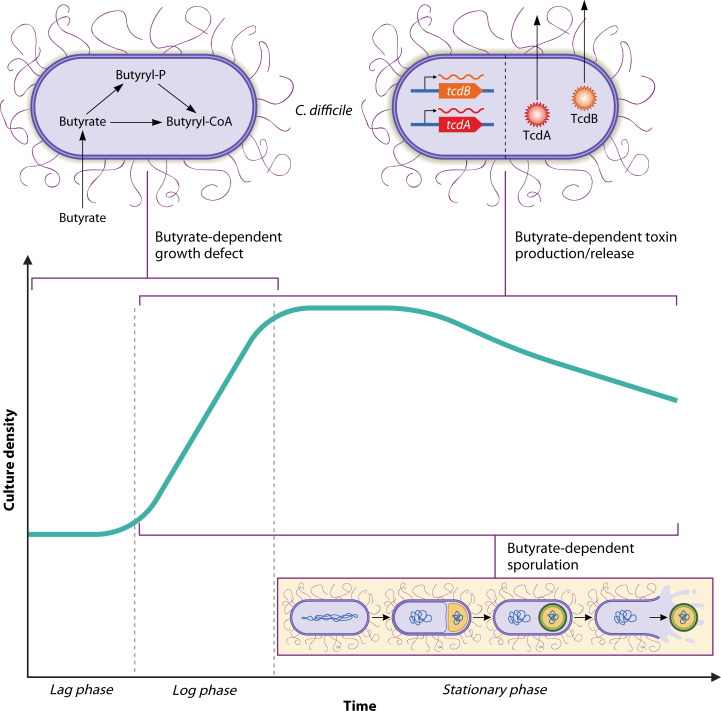 Fig 1
Fig 1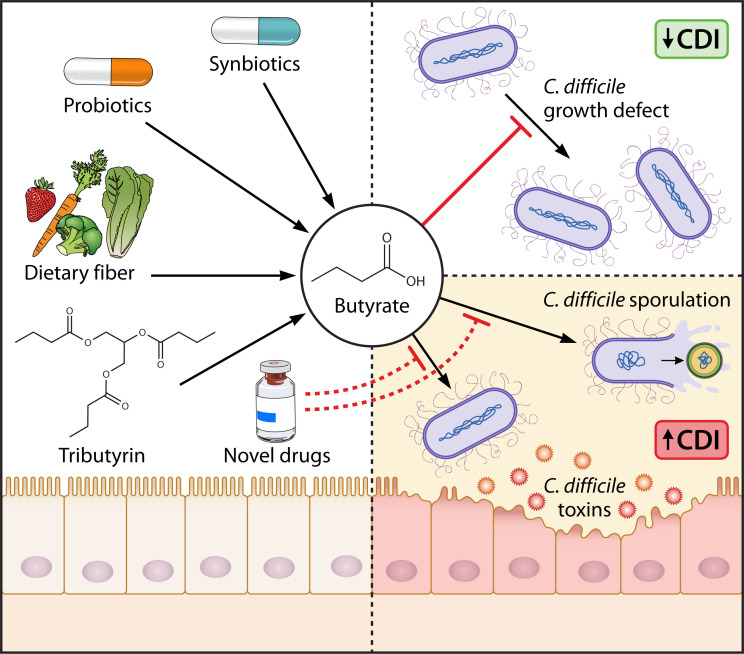 Fig 2
Fig 2| Reference | Media | Butyrate concentrations | General findings | |
|---|---|---|---|---|
| ( | Brain-heart infusion (BHI) | TTU 614 | 1.5 | The MIC of butyrate is 6.2 |
| ( | BHI | 1551 | 25 mM |
Butyrate inhibits No dose-dependent effect observed. |
| ( | Cartman’s | 630 | 10 mM | Butyrate decreases |
| ( | BHI | VPI 10463 | 1 mM | Butyrate negatively impacts |
| ( | Modified reinforced clostridial medium (mRCM) | 630 | 6.25 mM | Butyrate has dose-dependent impacts on |
| ( | BHI | 630 | 5 mM |
In BHI, butyrate has a dose-dependent growth defect in BHI in Supplementation of various sugars in CDMM modulates the butyrate-dependent growth defect: Medium supplemented with lactose and raffinose maintained the growth defect Medium supplemented with cellobiose, maltose, and trehalose had no butyrate-dependent growth defect Medium supplemented with fructose, mannose, and mannitol had butyrate-dependent growth enhancement. |
| ( | mRCM | 630 | 50 mM |
A butyrate-dependent growth defect is observed in 630 in an undefined complex medium (mRCM) but not in a defined minimal medium (BDM). The butyrate-dependent growth defect increases in conjunction with nutrient availability. The butyrate-dependent growth defect occurs in a medium where Supplementation with 13C4-butyrate revealed that butyrate is internalized into CoA pools and forces butyrogenic metabolic pathways in a reverse, metabolically unfavorable, direction. |
| Reference | Media | Butyrate concentrations | General findings | |
|---|---|---|---|---|
| ( | Peptone-yeast (PY) ± 10 mM cysteine | VPI 10463 | 30 mM |
Butyrate increases detectable toxin yield across all growth media tested. Butanol decreases toxin yield in a dose-dependent manner in PY. |
| ( | Cartman’s | 630 | 10 mM | Butyrate increases TcdB in culture supernatants in a dose-dependent fashion. |
| ( | Brain-heart infusion (BHI) | VPI 10463 | 1 mM | Butyrate (10 mM and 50 mM) increases TcdA and TcdB in culture supernatants. |
| ( | BHI | 630 | 5 mM |
Butyrate increases detectable toxins in 25 mM butyrate induces more toxin than 5 mM butyrate, but a significant dose-dependent response was not identified with an expanded range of concentrations. Elevated toxin levels are detectable as early as 6–12 h post-inoculation. Small but significant upregulation of Small but significant upregulation of |
| ( | Modified reinforced clostridial medium | 630 | 50 mM |
Butyrate positively impacts toxin levels in culture supernatants at 48 h post-inoculation. Butyrate does not strongly impact toxin transcripts ( Butyrate positively affects transcripts of Butyrate positively impacts extracellular lactate dehydrogenase at 48 h post-inoculation, leading to the hypothesis that butyrate impacts toxin release (potentially via autolysis). |
| Reference | Media | Butyrate concentration(s) | General findings | |
|---|---|---|---|---|
| ( | Brain-heart infusion | 630 | 5 mM |
Butyrate-dependent sporulation occurs in Butyrate-dependent sporulation occurs in Numerous sporulation genes are upregulated in the presence of 25 mM butyrate in BHI (e.g., stage II, III, IV, and V sporulation genes and sporulation sigma factors E, F, and G). |
| ( | Modified reinforced clostridial medium | 630 | 50 mM |
Numerous sporulation genes are upregulated in 630 in the presence of 50 mM butyrate in mRCM (e.g., stages II, III, IV, and V sporulation genes and sporulation sigma factors E, F, G, and K). Butyrate-dependent sporulation occurs in 630 and Butyrate does not increase viable spore counts in 630 grown in 70:30 medium. |
- —National Science Foundationhttp://dx.doi.org/10.13039/501100008982
- —Judy L. and Sal A. Troia Professorship in Gastrointestinal Disease Research
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMarine Sponges and Natural Products
CLOSTRIDIOIDES DIFFICILE IS A SERIOUS HEALTHCARE CONCERN
The Centers for Disease Control and Prevention classifies Clostridioides difficile as an urgent threat to the nation’s health, as it causes 450,000 infections, 15,000 deaths, and 1 billion dollars in excess healthcare costs per year in the United States (1, 2). Most C. difficile infections (CDIs) occur in healthcare settings, where CDI is the most common cause of infectious diarrhea (3). Known and suspected risk factors for CDI include antibiotics, proton pump inhibitors, impaired immune function, advanced age, and diet, all of which are associated with dysbiotic gastrointestinal (GI) microbiomes (4–6). Though most CDIs are associated with antibiotic treatment, 22% of individuals with community-acquired CDI have no recent history of antibiotic use. Factors affecting persistent and recurrent CDIs remain poorly defined (7, 8). Despite the morbidity and mortality caused by C. difficile, up to 15% of healthy adults are asymptomatic carriers of toxigenic C. difficile (9), highlighting the gaps in our understanding of C. difficile.
C. difficile pathogenesis is mediated by secreted protein toxins. The two main toxins produced by C. difficile are TcdA and TcdB. C. difficile releases these toxins into the gut lumen. They are internalized into colonocytes and inactivate Rho GTPases, which leads to disruption of the actin cytoskeleton and tight junctions. Toxin-induced cellular damage causes cell lysis, inflammation, and a weakened epithelial barrier, which lead to the diarrhea characteristic of CDI (10–13). tcdA and tcdB are encoded within a 19.6 kb pathogenicity locus (14), which also contains three accessory genes involved in toxin regulation and secretion. Specifically, tcdR encodes the alternative sigma factor responsible for activating the expression of toxin genes (15), tcdC encodes the anti-sigma factor that inhibits TcdR (16), and tcdE encodes a holin-like protein that forms pores in the C. difficile cell membrane and is involved in toxin secretion (17, 18). Some “hypervirulent” C. difficile strains produce a third secreted toxin, CDT, which is internalized into colonocytes and catalyzes ADP ribosylation; this activity disrupts the actin cytoskeleton and contributes to CDI pathogenesis (19–21). Emerging evidence suggests that C. difficile toxin-mediated inflammation benefits C. difficile by allowing it to acquire host nutrients (22, 23) and outcompete inflammation-sensitive gut microbes (22–25).
Another key feature of C. difficile biology is its ability to form spores, likely in response to stress and nutrient limitation. Sporulation in C. difficile has been tied to its metabolic environment, particularly with emphasis on peptides (26), carbon catabolite availability (27, 28), and branched amino acids present in its environment (29). Genetic regulation of C. difficile sporulation is tightly controlled (26, 30, 31). Through a complex series of morphological and biochemical changes, sporulating C. difficile cells divide asymmetrically and form an environmentally resistant spore, which is released from the larger mother cell by cell lysis. Spores are highly resistant to environmental stresses such as heat, chemicals, desiccation, oxygen, and ethanol-based sanitizers (32). These spores allow C. difficile to survive outside the GI tract and transmit to new hosts, where they germinate to form vegetative cells (33).
THE MICROBIAL AND METABOLIC MILIEU OF CDI
Dysbiosis is the primary risk factor for CDI, and several microbial taxa directly impact C. difficile fitness (34–36). However, inter-individual variation in gut microbiome composition complicates definitions of CDI susceptibility and resistance. Human studies and animal models suggest that microbiome-dependent metabolites, rather than microbiome composition, define CDI susceptibility and resistance (37–40). Therefore, a focus on metabolites may allow for more readily translatable findings.
The gut contains thousands of diverse molecules derived from the diet and host/microbiome metabolism. Many of these impact C. difficile fitness and pathogenesis. For example, bile acids, metals, amino acids, sugars, organic acids, and short-chain fatty acids (SCFAs) affect C. difficile in vitro and in animal models (22, 23, 35–37, 41–46). SCFAs (in particular, acetate, propionate, and butyrate) are major metabolic products of microbiome metabolism and are primarily generated by microbial degradation of dietary fiber (47, 48). Collectively, SCFAs reach concentrations >100 mM in the colon and are the most concentrated metabolites in the human distal gut (49). SCFAs are influenced by microbiome composition, diet, antibiotics, and inflammation (50). Most previous research on SCFAs has focused on their impacts on the host. For example, SCFAs are metabolized by colonocytes or are transported systemically via portal circulation (47). SCFAs also enhance gut barrier function and modulate immune responses (47, 48). SCFA deficiencies are associated with inflammatory bowel disease and increased susceptibility to pathogens (25). Because SCFAs are so pivotal in host-microbiome cross-talk, microbiome studies often equate a “healthy gut” with high SCFAs and a “dysbiotic gut” with low SCFAs (25).
Of the SCFAs produced in the GI tract, butyrate stands out as an impactful microbiome-host co-metabolite. Microbiome-produced butyrate is the main source of energy for gut colonocytes (51, 52), where it is taken up and consumed by colonocytes through fatty acid β-oxidation, a process that consumes oxygen and supports the growth of obligate anaerobes. When butyrate is limited, colonocytes switch from fatty acid β-oxidation to glycolysis. Glycolysis does not consume oxygen and subsequently contributes to dysbiosis (53, 54). Additionally, butyrate promotes the integrity and maintenance of gut barrier function by increasing the number of tight junctions between colonocytes (55), increasing expression of Mucin 2 to strengthen the mucus layer of the gut (56), and by enhancing HIF-1 activation in colonocytes (57). Additionally, butyrate has direct immunomodulatory effects. It suppresses proinflammatory effectors in macrophages within the lamina propria (58), as well as the differentiation of dendritic cells from bone marrow stem cells (59) through histone deacetylase inhibition.
In addition to its direct effects on the host, animal and human data show that high butyrate characterizes a gut that is non-permissive to CDI (37, 45, 60, 61). In vitro, butyrate inhibits C. difficile growth, increases sporulation, and leads to elevated C. difficile toxins in culture supernatants (37, 45, 46, 57, 60, 62–64). Additionally, exogenous butyrate is internalized into C. difficile cells and is incorporated into its butyrogenic metabolic pathways, potentially contributing to these phenotypes. The conclusion that butyrate is associated with non-permissive environments for C. difficile in vivo while also enhancing C. difficile’s virulence mechanisms in vitro seems contradictory. However, taken together, these data on CDI permissiveness, growth, sporulation, and toxin expression/release suggest that C. difficile senses butyrate as a signal of a competitive environment and adjusts its virulence to maintain a dysbiosis-associated niche or to transmit to new hosts (25). Therefore, current data suggest that butyrate is a context-dependent regulator of C. difficile fitness and virulence traits. As such, butyrate stands out as an impactful target to better understand microbiome-host co-metabolism and to improve therapies against C. difficile.
THE IMPACTS OF FIBER CONSUMPTION AND GI BUTYRATE ON CDI IN MICE AND HUMANS
A growing body of literature suggests that fiber-deficient diets favor infection by bacterial pathogens such as C. difficile in humans and in animal models of CDI (37, 45, 65–68). We previously showed that fiber-deficient rodent diets (which recapitulate fiber deficiencies of human Western diets) perpetuate CDI in mice. In contrast, mice fed high-fiber diets containing a complex mixture of fiber types (standard rodent chow) (37) or diets containing isolated fiber types that elevate GI butyrate (e.g., inulin or resistant maltodextrin) (45) clear C. difficile within days. Similarly, dietary xanthan gum leads to rapid clearance of C. difficile from the mouse gut and is associated with elevated GI butyrate levels (69). In contrast, fiber types that do not elevate GI butyrate (e.g., fructooligosaccharides and gum Arabic) do not suppress C. difficile burdens (45). These data suggest that CDI suppression is not generalizable to all fibers, but rather to a fiber’s butyrogenic capacity. To date, eight diets containing various fiber types confirm this link (45, 69), but it is unclear whether the effects are caused by butyrate or are simply associated with elevated butyrate (70).
Additional connections between butyrate and C. difficile fitness have been drawn using mouse and hamster models of CDI, without manipulating dietary fiber. In a Syrian hamster model of CDI, both C. difficile colonization and symptomatic CDI differed between infant vs adult animals, where the development of the hamster gut microbiome and subsequent production of SCFAs were correlated with these observations. From this study, only GI butyrate levels in the hamster reached inhibitory levels for C. difficile, indicating butyrate’s importance during experimental infection (60). In another study using gnotobiotic mice colonized with a butyrate producer (Clostridium sardiniense), mice succumbed more rapidly to CDI than gnotobiotic mice colonized with an amino acid fermenter (Paraclostridium bifermentans), highlighting context-dependent effects of butyrate on CDI (71). Finally, SCFAs have beneficial pleiotropic effects on host biology, but the impacts of SCFAs on the host as they relate to CDI outcomes are mostly unexplored. One study treated mice with butyrate and showed that they had less severe CDI but did not measure C. difficile burdens (70). These effects were mediated through farnesoid X receptor, which is activated by bile acids, but not by butyrate (70). Also, rather than treating with tributyrin, a prodrug of butyrate that efficiently delivers butyrate to the distal GI (72, 73), mice were given butyrate by oral gavage, which is absorbed by the proximal GI (74). So, it is unclear if the changes in this study were due to systemic effects of butyrate on the immune system. Another study administered both butyrate and tributyrin to mice infected with C. difficile and found that while CDI severity was lessened in vivo, butyrate/tributyrin did not impact colonization resistance against C. difficile or its toxin production (using both germ-free mice and conventional mice) (57). These findings suggest context-dependent differences in vivo, which can drastically change how butyrate impacts CDI, emphasizing that more knowledge of the direct effects and relative impacts of fiber and butyrate on C. difficile fitness and virulence phenotypes during CDI is needed.
There are similar links between fiber, butyrate, and CDI in humans. We showed that CDI patients have lower butyrate (but not acetate or propionate) in their stool than healthy controls (45). Others showed that CDI patients have lower propionate and butyrate (but not acetate) than healthy controls (70), and GI butyrate is positively associated with successful fecal transplant for recurrent CDI (61). Also, fiber consumption is associated with lower odds of C. difficile colonization in humans and a concomitant increase in GI SCFAs (67, 68). Therefore, fiber consumption and high GI butyrate are associated with lower CDI risk in humans.
Diet has a much broader impact than simply modulating GI butyrate levels. Shifts in diet lead to large-scale changes to gut microbiome membership (37), changes in host metabolism (51, 52), and impact other parameters such as stool consistency and gut transit time, all indicating that the effects of butyrogenic fiber on CDI are also impacted by additional variables other than butyrate. Since existing in vivo work occurred against the backdrop of the complex microbial and metabolic milieus of the GI tract, a more granular understanding of butyrate production in the gut and its direct impacts on C. difficile is needed.
THE EFFECTS OF EXOGENOUS BUTYRATE ON C. DIFFICILE GROWTH AND METABOLISM
Butyrate inhibits the growth of diverse C. difficile isolates in vitro (37, 45, 46, 57, 60, 62, 63). The presence and magnitude of butyrate-dependent growth inhibition vary based on concentrations of butyrate and other nutrients in the media (Table 1). For example, while several studies identified a dose-dependent inhibition of growth with butyrate supplementation, others were unable to replicate those findings. In one study, the addition of various sugar sources, such as cellobiose, maltose, and trehalose, abolished the butyrate-dependent growth defect, and fructose, mannose, and mannitol supplementation induced a butyrate-dependent increase in growth (63). This indicates that differences in nutrient availability drive differences in metabolic flux or preferences for different metabolic pathways that may influence butyrate’s inhibitory effects on C. difficile. Additionally, a study from our group identified that in a basal-defined medium (BDM), C. difficile does not produce its own butyrate and does not experience a butyrate-dependent growth defect (46), further emphasizing that metabolic flux through butyrogenic pathways is important for this phenotype. Supportively, when supplied with exogenous butyrate, C. difficile produces less of its own butyrate. To address the underlying effects of exogenous butyrate on C. difficile butyrogenic metabolism, we supplemented cultures with ^13^C_4_-butyrate. We found that ^13^C_4_-butyrate is incorporated into intracellular CoA pools where it is metabolized in an energetically unfavorable direction to crotonyl-CoA and hydroxybutyryl-CoA (46). This demonstrates two important concepts. First, exogenous butyrate is internalized into C. difficile cells and alters C. difficile metabolism. Second, butyrate-dependent growth defects only occur when C. difficile is producing butyrate as a metabolic end product.
However, butyrate does not inhibit the growth of all bacteria (75, 76). One study on Bacteroides identified that while butyrate can have an inhibitory effect, supplementation with different sugar sources can modulate and even negate this effect depending on the strain used (76). Therefore, different gut microbes differentially sense and respond to butyrate, possibly highlighting important differences in their metabolic capabilities and their niches within the gut. It is reasonable to assume that organisms that are negatively affected by butyrate respond to butyrate-rich environments by altering their gene expression. In the case of a pathogen like C. difficile, it is likely that these responses involve the expression of genes involved in virulence.
BUTYRATE-DEPENDENT EFFECTS ON C. DIFFICILE TOXINS
During dysbiosis, C. difficile can access nutrients otherwise consumed by the microbiome, and it proliferates. However, as the microbiome recovers from dysbiosis, C. difficile must compete with these microbes for nutrients (24). C. difficile can leverage the inflammation created by its toxins to enhance its infection by both suppressing the growth and recovery of other gut microbes (23, 77) and liberating host metabolites (23) that provide this pathogen with a competitive advantage. Supportively, C. difficile toxin expression and release are directly tied to multiple features of a competitive gut environment (e.g., low proline, branched-chain amino acids, fructose-1,6-bisphosphate, or cysteine; and high autoinducing peptides or butyrate) (78). This highlights that the exquisite control C. difficile has over its virulence. We and others showed that butyrate leads to increases in C. difficile toxins in culture supernatants in a dose-dependent fashion (37, 46, 57, 63, 64). As observed for butyrate-dependent growth inhibition in C. difficile, the presence and magnitude of butyrate-dependent toxin expression and release vary based on the concentrations of butyrate and other nutrients in the media (Table 2). Nonetheless, the consensus from these studies confirms that butyrate-induced toxin production/release occurs across diverse C. difficile strains and under different growth conditions, emphasizing the generalizability of the phenotype.
Two recent studies interrogated butyrate-dependent expression of toxin genes and toxin release machinery. In our work, RNA-seq revealed that butyrate does not strongly impact tcdA/tcdB transcription in C. difficile. While tcdA/tcdB transcripts are slightly elevated during log phase growth with butyrate, these increases are less than a twofold increase (log_2_ fold change <1), and therefore, these genes were deemed not strongly differentially expressed in our work. Instead, tcdE, CD630_21840 (a putative endolysin that facilitates toxin release independent of autolysis, potentially in conjunction with TcdE) (79), and cwp19 (a lytic transglycosylase involved in stationary phase autolysis) (80) are all strongly upregulated (log_2_ fold change >1, adjusted P-value < 0.05) during the stationary phase, coinciding with the high toxin levels in culture supernatants. Cwp19-mediated autolysis and TcdE-mediated secretion are co-existing mechanisms for toxin release (80). Supportively, butyrate exposure leads to increased autolysis of C. difficile cells (46). These data suggest that butyrate affects TcdE-dependent secretion and Cwp19-dependent autolysis, which together contribute to butyrate-dependent toxin release. However, it is unclear if toxin release via autolysis and TcdE occurs on different time scales or due to different signals in culture and during CDI. Another study that grew C. difficile in a different growth medium (brain-heart infusion [BHI]) showed butyrate-dependent increases in toxin levels in culture supernatants. This corresponded to small but significant increases in tcdR and tcdA transcripts and small but significant decreases in tcdC transcripts in the presence of butyrate (63). Taken together, these data support that different metabolic landscapes may differentially impact butyrate-dependent toxin expression and release.
BUTYRATE IMPACTS C. DIFFICILE SPORULATION
Spores are the sole vehicle of C. difficile transmission. While sporulation has been previously tied to metabolite availability, butyrate-responsive sporulation is a newly appreciated facet of C. difficile biology that ties C. difficile transmission to yet another aspect of the metabolic activity of the gut microbiome (46, 63) (Table 3).
While the mechanisms underlying butyrate-dependent sporulation in C. difficile are unknown, this new literature, combined with previous work in C. difficile and relatives, provides impactful starting points. Notably, in both C. difficile and Bacillus subtilis, sporulation initiation is controlled by phosphorylation of the master regulator, Spo0A. Once phosphorylated, Spo0A is responsible for inducing expression of numerous genes, including the early-stage sporulation sigma factors, SigE and SigF. These early-stage sporulation sigma factors then activate transcription of the genes encoding late-stage sporulation sigma factors SigG and SigK, in a hierarchical cascade of gene expression to carry out this complex biological process (81). RNA-seq from two independent studies showed that the most prominent class of genes upregulated in response to butyrate is “sporulation.” Specifically, in one study by Baldassare et al., butyrate induced expression of sporulation sigma factors such as SigE, SigF, SigG, as well as other stages II, III, IV, and V sporulation genes and endopeptidases (63). This group also identified that genes encoding many of the known regulators of virulence and sporulation, such as CcpA, Rex, PrdR, and CodY, were not significantly differentially expressed in the presence of butyrate. Supportively, in our work, butyrate also induced expression of genes encoding sporulation sigma factors, SigE, SigF, SigG, and SigK, and numerous stages II, III, IV, and V sporulation genes and additional spore-specific genes (46). In contrast to the paper by Baldassare et al., our work showed significant (adjusted P-value < 0.05) downregulation of ccpA, rex, prdR, and codY; however, these were small changes in expression (and log_2_-fold change <2 and >−2) and were therefore not reported as differentially regulated. Upregulation of the downstream sporulation sigma factors but not spo0A during log-phase growth suggests that butyrate could be directly or indirectly impacting Spo0A activity to trigger sporulation initiation and transcription of these sigma factors; however, additional investigation into the molecular and genetic mechanisms underlying this phenotype is necessary.
Despite similar conclusions between these two studies, some differences in phenotypes were observed, which might help to hone future approaches for the mechanistic dissection of butyrate-dependent sporulation. For example, our work did not detect elevated spore counts during log phase growth (46), while Baldassare et al. did (63). In addition, our work was unable to show that butyrate increased spore counts using 70:30 medium (46), while Baldassare et al. did (63). These discrepancies could be due to differences in media types used (modified reinforced clostridial medium [mRCM] vs BHI) or differences in the methods of detection for the 70:30 medium. Both groups used the same strain and medium (70:30); however, this difference in spore quantification methods (phase contrast microscopy, which does not assay spore viability, vs culturing viable spores) may have led to the conflicting results, as spores must be germination-proficient to be detected via culturing. Regardless, the detection of increased sporulation by butyrate in BHI and mRCM, two media known for low sporulation efficiency, from two separate research groups, indicates the importance of butyrate-rich environments in initiating sporulation in C. difficile. Furthermore, this work highlights the utility of diverse in vitro growth conditions to define nodes within the complex regulatory network that controls C. difficile sporulation.
DISTINCT PHENOTYPES OR AN INTEGRATED RESPONSE?
Butyrate exposure leads to three well-documented phenotypes in C. difficile: growth inhibition, increased toxin production/release, and enhanced sporulation. These phenotypes could reflect an integrated physiological response mediated by to-be-determined regulatory mechanisms. CodY and CcpA, which sense nutrient status, simultaneously repress toxin and sporulation genes in nutrient-rich environments (27, 29). RstA drives sporulation but dampens toxin expression and motility (82). If butyrate impacts the activity of one or more of these regulators, either directly or via intracellular metabolites (such as butyryl-CoA) that accumulate during butyrate exposure, it could lead to co-regulation of these phenotypes ([Tables 1 to 3](#T1 T2 T3); Fig. 1). However, the known modes of sporulation and toxin co-regulation in C. difficile impact transcript levels of both spo0A and the toxin genes. Therefore, due to the absence of strong butyrate-dependent changes to tcdA, tcdB, and spo0A transcripts in existing studies, it is reasonable to hypothesize that these phenotypes are regulated by distinct and possibly novel mechanisms.
Timing of butyrate’s effects on C. difficile biology in vitro. Three butyrate-dependent phenotypes are observed in rich growth media. A butyrate-dependent growth defect is observed during log-phase growth, while both sporulation and toxin phenotypes are observed starting during log-phase growth and into stationary-phase growth, depending on experimental conditions. The specialized callouts highlight current understanding and existing gaps in knowledge of these phenotypes. The butyrate-dependent growth defect coincides with internalization of exogenous butyrate and accumulation of intracellular butyryl-CoA and downstream metabolic intermediates in C. difficile’s butyrate production pathways. Butyrate-dependent toxin gene expression and toxin release are likely involved in the toxin phenotype. Butyrate-dependent sporulation has been observed; however, little is known regarding how this occurs on a molecular level. Additional research is necessary to further unravel the mechanism(s) underlying all three of these phenotypes. Artwork courtesy of Patrick Lane, ScEYEnce Studios; reprinted with permission.
Alternatively, and in support of segregation of these phenotypes, C. difficile can partition into distinct sub-populations, where some cells express toxin, others sporulate, and a minority do both (83). The physical environment (e.g., solid vs liquid media) appears to reinforce this division of labor. In this context, butyrate may act as a differential signal to modulate behavior in a subset of cells based on local butyrate concentration, intracellular redox status, or proximity to other microbes. In line with this idea, the concentration of butyrate matters. Dose-dependent effects on butyrate-dependent phenotypes have been observed ([Tables 1 to 3](#T1 T2 T3)). While cecal and colonic butyrate levels can reach ~25 mM (49, 84), bulk measurements do not account for concentration gradients that exist due to the spatial organization of producers/consumers (85). A C. difficile cell adjacent to a strong butyrate-producing organism may experience a drastically different environment than one that is several microns or even millimeters away. So, while some previous work to uncover butyrate-dependent effects on C. difficile used “super-physiological” concentrations of butyrate, we feel that this choice is appropriate due to these limitations and as a tool to identify and dissect phenotypes, especially if dose-dependent effects are observed. So, the sum of these effects could have varying impacts on C. difficile’s responses to butyrate, especially if phenotypes are sensitive to different threshold concentrations. Taken together, there is an unmet need for further dissection of the molecular, genetic, metabolic, and ecological underpinnings of these phenotypes.
POSSIBLE BENEFITS AND RISKS OF THERAPEUTIC MANIPULATION OF BUTYRATE DURING CDI
From a therapeutic standpoint, butyrate-dependent growth inhibition, butyrate-dependent modulation of the immune system, and observations that butyrate-rich gut environments exclude C. difficile are encouraging. This suggests that therapeutic elevation of butyrate in the GI tract could be useful in mitigating CDI in at-risk human populations. However, in addition to these effects, emerging evidence supports that butyrate is a cue exploited by C. difficile to time virulence and transmission. Butyrate-dependent elevation of toxins and spores suggests that C. difficile may sense rising butyrate levels as a signal of an inhospitable environment, where competing microbes are in abundance or one that is at carrying capacity for C. difficile. Either way, increased sporulation would enhance transmission to new hosts. Likewise, elevations in C. difficile toxins increase inflammation, which may slow host recovery, reduce the fitness of inflammation-sensitive microbes with which C. difficile competes for nutrients, and further aid in transmission. Butyrate-dependent effects on these virulence traits support the hypothesis that butyrate acts as a signaling molecule for C. difficile, akin to those used in quorum sensing.
In a complex host-associated microbial ecosystem like the gut, which of these butyrate-dependent effects is dominant? Previous work showed that switching mice from a fiber-deficient to a fiber-rich diet (characterized by low GI butyrate and high GI butyrate), C. difficile burdens are suppressed, but there is a transient increase in detectable toxins in the stool, which correlates with a transient increase in GI inflammation (37). In this case, it is reasonable to hypothesize that butyrate-dependent growth inhibition, butyrate-dependent effects on the host, and butyrate-independent effects of fiber dominate over the effects of toxin expression. However, as described above in section “The impacts of fiber consumption and GI butyrate on CDI in mice and humans,” there is emerging evidence that suggests that multiple variables can influence the balance of these effects.
Therefore, prior to translating these findings into therapeutic applications (Fig. 2), more basic research is needed. Fiber-based interventions are attractive because they are safe, accessible, and beneficial to host health. However, their success depends on microbiome composition, butyrate-independent effects of fiber (86–88), patient access to fiber-rich foods, and patient compliance. Synbiotics (the combination of probiotic organisms and prebiotics, non-digestible fibers which feed the probiotic organism) may overcome some of these complications and could be readily engineered based on known fiber-degrading and butyrate-producing probiotic organisms. Another option could be oral administration of butyrate or tributyrin. These strategies have shown efficacy in mice by protecting intestinal epithelial cells from C. difficile toxins through stabilization of HIF-1 (57). Alternatively, new drugs could be developed based on findings from ongoing research in this area. Specifically, small molecule inhibitors of key steps involved in butyrate-dependent regulation of toxins or sporulation could be developed, which could be administered to specifically inhibit butyrate-responsive virulence phenotypes.
Possible therapeutic strategies to elevate gastrointestinal butyrate or target butyrate-responsive virulence traits. Possible strategies of elevating intestinal butyrate include dietary fiber, butyrogenic probiotics/synbiotics, and tributyrin administration. Elevated butyrate has the potential to reduce C. difficile burdens but also increase the expression of virulence traits. Therapeutic induction of the butyrate-induced growth defect could decrease C. difficile proliferation and simultaneously offer other gut microbes a competitive nutritional advantage against C. difficile. However, therapeutic elevation of butyrate could also positively impact butyrate-induced toxin expression/release and sporulation, which would further exacerbate CDI symptoms and promote transmission to new hosts. Additional research is needed to identify the host-, microbiome-, and C. difficile-based determinants that affect the balance of these phenotypes in vivo, which also has the potential to inform the development of new drugs to minimize toxin and sporulation phenotypes and favor CDI resolution. Artwork courtesy of Patrick Lane, ScEYEnce Studios; reprinted with permission.
CONCLUSIONS AND FUTURE DIRECTIONS
Current data show that butyrate-rich gut environments are generally non-permissive to C. difficile colonization and disease. While butyrate negatively impacts C. difficile fitness in vitro, butyrate exposure also leads to elevations in C. difficile toxins and enhanced sporulation. These combined observations underscore the importance of butyrate as a modulator of gut ecosystem function and as a cue for C. difficile to regulate its virulence and transmission. While substantial progress has been made, critical questions remain. First, we lack a mechanistic understanding of whether butyrate’s growth inhibitory effects are a direct result of interference with intracellular CoA pools, altered redox balance, cytoplasmic acidification (89), or pushing butyrogenic reactions in an unfavorable direction. Comparisons of C. difficile with analogous literature on other organisms’ responses to SCFAs (75, 89) will help to provide additional future research directions, define clade-specific responses, and better understand how SCFAs may directly shape the microbiome in health and disease. Second, our work suggests that toxin release, rather than canonical transcriptional control of tcdA and tcdB, is the dominant toxin-related phenotype under butyrate exposure. Butyrate-dependent upregulation of genes like tcdE and cwp19, as well as increased autolysis, supports this view. However, it is unclear if toxin secretion and autolysis pathways act simultaneously or on different timescales or if these effects are driven by butyrate itself or downstream metabolites (e.g., butyryl-CoA). Third, butyrate exposure enhances C. difficile sporulation but without altering spo0A transcript levels. This suggests that novel regulatory pathways are also involved in this context. One possibility is that there is post-transcriptional or post-translational modification of key regulators like Spo0A, for example, through butyrylation (as has been observed in C. acetobutylicum [90]) or through Hfq-dependent sRNAs (91). Alternatively, butyrate may impact known global regulators or stress responses in unappreciated ways to affect sporulation. Additionally, understanding the concentrations of butyrate that represent the “threshold” for each of these phenotypes is an important piece of the puzzle that remains to be answered and will further contextualize these phenotypes in the complex microbial and metabolic milieus of the gut. All of these—including whether growth, sporulation, and toxin effects are coordinated through similar regulatory pathways, which of these effects dominate in vivo, and over what time frames—represent clear targets for future molecular and genetic dissection.
Taken together, this review highlights the utility of microbial metabolites as entry points to understanding pathogen biology. Continued efforts to understand how butyrate (and other host and microbiome-derived signals) is sensed by C. difficile will yield a better understanding of its pathogenesis and perhaps inform strategies to mitigate CDI with minimal impacts to our microbiomes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, Mc Donald LC. 2015. Burden of Clostridium difficile infection in the United States. N Engl J Med 372:825–834. doi:10.1056/NEJ Moa 140891325714160 PMC 10966662 · doi ↗ · pubmed ↗
- 2Guh AY, Mu Y, Winston LG, Johnston H, Olson D, Farley MM, Wilson LE, Holzbauer SM, Phipps EC, Dumyati GK, Beldavs ZG, Kainer MA, Karlsson M, Gerding DN, Mc Donald LC, Emerging Infections Program Clostridioides difficile Infection Working Group. 2020. Trends in U.S. burden of Clostridioides difficile infection and outcomes. N Engl J Med 382:1320–1330. doi:10.1056/NEJ Moa 191021532242357 PMC 7861882 · doi ↗ · pubmed ↗
- 3Johnson S, Gerding DN. 1998. Clostridium difficile--associated diarrhea. Clin Infect Dis 26:1027–1034. doi:10.1086/5202769597221 · doi ↗ · pubmed ↗
- 4Bignardi GE. 1998. Risk factors for Clostridium difficile infection. J Hosp Infect 40:1–15. doi:10.1016/s 0195-6701(98)90019-69777516 · doi ↗ · pubmed ↗
- 5Di Bella S, Sanson G, Monticelli J, Zerbato V, Principe L, Giuffrè M, Pipitone G, Luzzati R. 2024. Clostridioides difficile infection: history, epidemiology, risk factors, prevention, clinical manifestations, treatment, and future options. Clin Microbiol Rev 37:e 0013523. doi:10.1128/cmr.00135-2338421181 PMC 11324037 · doi ↗ · pubmed ↗
- 6Eeuwijk J, Ferreira G, Yarzabal JP, Robert-Du Ry van Beest Holle M. 2024. A systematic literature review on risk factors for and timing of Clostridioides difficile infection in the United States. Infect Dis Ther 13:273–298. doi:10.1007/s 40121-024-00919-038349594 PMC 10904710 · doi ↗ · pubmed ↗
- 7Khanna S, Pardi DS, Aronson SL, Kammer PP, Baddour LM. 2012. Outcomes in community-acquired Clostridium difficile infection. Aliment Pharmacol Ther 35:613–618. doi:10.1111/j.1365-2036.2011.04984.x 22229532 PMC 3293482 · doi ↗ · pubmed ↗
- 8Abou Chakra CN, Pepin J, Sirard S, Valiquette L. 2014. Risk factors for recurrence, complications and mortality in Clostridium difficile infection: a systematic review. P Lo S One 9:e 98400. doi:10.1371/journal.pone.009840024897375 PMC 4045753 · doi ↗ · pubmed ↗