Detection and genetic characterization of Uukuvirus lihanense (Uukuvirus, Phenuiviridae) in hard ticks from the Colombian Caribbean
Ketty Galeano, Yesica López, Camilo Guzmán, Yeimi López, Héctor Contreras, Alejandra Garcia, Luis Romero, Caty Martínez, Daniel Echeverri, Luis Paternina, Alfonso Calderón, German Arrieta, Salim Mattar

TL;DR
Researchers found Uukuvirus lihanense in ticks from the Colombian Caribbean and compared its genetic makeup to other tick-borne viruses.
Contribution
First report of Uukuvirus lihanense in Amblyomma dissimile ticks and genetic characterization in the region.
Findings
Uukuvirus lihanense was detected in three tick species from the Colombian Caribbean.
The virus sequences were phylogenetically related to those from other regions in the Americas.
This is the first report of the virus in Amblyomma dissimile ticks.
Abstract
Ticks are arthropod vectors that transmit pathogens important to human and animal health. The objective of this work was to identify Uukuvirus lihanense in the metatranscriptome of hard ticks. Between October 2022 and June 2023, ticks were collected from rural areas of the Colombian Caribbean area of the departments of Córdoba and Cesar. High-throughput sequencing (next-generation sequencing) was performed using MGI’s DNBSEQ-G50RS. Bioinformatics analyses were performed in Galaxy, diamond and IQ-TREE2. A total of 766 ticks were collected; 87.33% (669/766) were Rhipicephalus microplus, 5.4% (42/766) Dermacentor nitens, 4.2% (32/766) Rhipicephalus sanguineus and 3.0% (23/766) Amblyomma dissimile. Complete and partial L and S segments of Uukuvirus lihanense (LITV) were detected in the metatranscriptome of A. dissimile, D. nitens and R. microplus. The LITV sequences found are…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
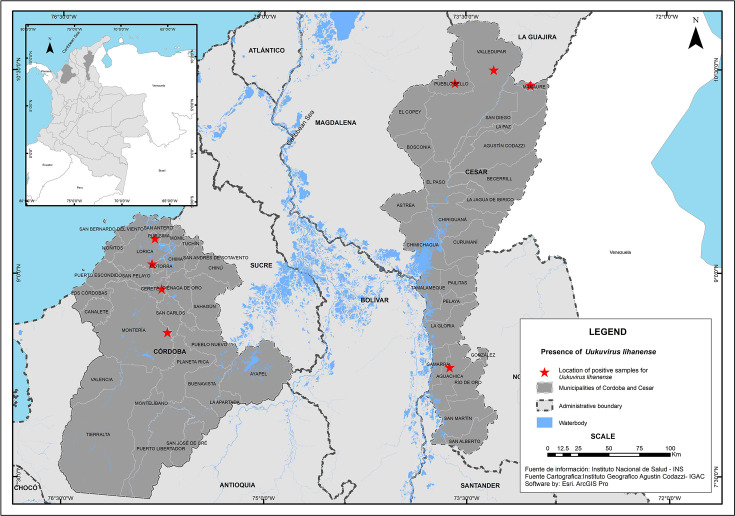 Fig. 1
Fig. 1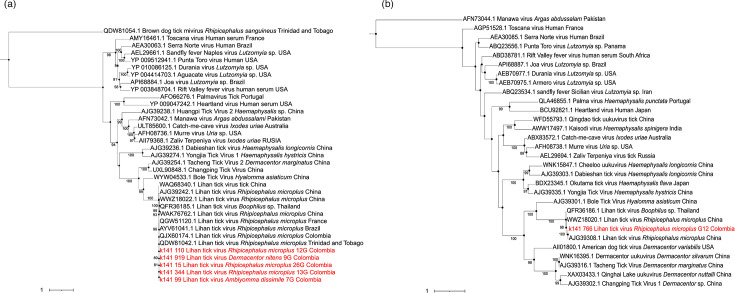 Fig. 2
Fig. 2| Code | Species | Sex/stage | Host | Location | Total reads | Total base (bp) | Contig | Segments of LITV | ||
|---|---|---|---|---|---|---|---|---|---|---|
| L | S | Segment | Segment S-nucleocapsid | |||||||
| 7G |
| ♀ | Iguana | Cesar | 30.702.552 | 4.593.014 | 1.993 | 1.304 aa | ||
| 9G |
| ♀ | Bovine | Cesar | 48.847.884 | 7.303.275 | 4.854 | 1.576 aa | ||
| 11G |
| ♀ | Bovine | Cesar | 36.841.332 | 4.981.765 | 3.337 | 53 aa | ||
| 12G |
| ♂ | Bovine | Cesar | 29.863.708 | 4.461.170 | 6.478 | 726 | 2.151 aa | 146 aa |
| 13G |
| N | Bovine | Cesar | 33.981.760 | 5.056.211 | 6.486 | 2.151 aa | ||
| 26G |
| ♀ | Bovine | Córdoba | 36.507.216 | 4.970.865 | 1.874 | 511 aa | ||
| 28G |
| ♀ | Dog | Córdoba | 30.810.816 | 4.096.377 | ||||
| 29G |
| ♂ | Bovine | Córdoba | 42.137.144 | 5.907.281 | 1813 | 53 aa | ||
| 117G |
| N | Bovine | Córdoba | 27.668.692 | 3.708.193 | 873 | 53 aa | ||
- —http://dx.doi.org/10.13039/100007637 Departamento Administrativo de Ciencia, Tecnología e Innovación (COLCIENCIAS)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsViral Infections and Vectors · Vector-Borne Animal Diseases · Mosquito-borne diseases and control
Data Summary
The authors confirm all supporting data, code and protocols have been provided within the article. Sequences are available in the Sequence Read Archive with accession nos. SRX25381865, SRX25381866, SRX25381867, SRX25381868 and SRX25381869.
Author Statement
The brief report submitted entitled, ‘Detection and genetic characterization of Uukuvirus lihanense (Uukuvirus, Phenuiviridae) in hard ticks from the Colombian Caribbean’ and the previously published brief report ‘Hard ticks (Ixodida: Ixodidae) in the Colombian Caribbean harbour the Jingmen tick virus: an emerging arbovirus of public health concern’. They are part of the same research project titled ‘Fortalecimiento de las capacidades de investigación con relación a las enfermedades transmitidas por vectores de las universidades de Córdoba y Cesar 2020–2023 en Córdoba, Cesar’.
Introduction
Ticks are ectoparasites widely distributed globally and are known as vectors of many viruses important to human and animal health [13]. These arthropods host a variety of negative-sense RNA viruses, many of them emerging pathogens, such as the severe fever with thrombocytopenia syndrome virus (Bandavirus dabieense) [4] and re-emerging ones such as Crimean-Congo haemorrhagic fever virus, which have caused outbreaks and fatal cases in humans [5].
The genus Uukuvirus belongs to the family Phenuiviridae [610], where critical human pathogens are found, such as B. dabieense and Bandavirus heartlandense discovered in China and the USA, respectively [1112]. The genome of Uukuviruses is a negative-sense segmented RNA, with three segments (L, M, S) encoding four proteins. The RNA-dependent RNA polymerase (L), external glycoproteins (Gn and Gc), a nucleocapsid protein (S) and a nonstructural protein (NSs) [610]. However, Uukuvirus lihanense (LITV) lacks the M segment, a critical component that allows cell entry. This absence of the M segment in LITV is significant as it affects the virus’s ability to infect cells, potentially influencing its pathogenicity and transmission dynamics [5].
Given the vast array of viruses found in ticks, the advent of next-generation sequencing (NGS) technology has revolutionized our ability to investigate and identify tick viromes [3,1315]. In Colombia, NGS-based studies have recently unveiled the diversity of viruses in ticks, including the genera Orthoflavivirus, Orthonairovirus, Bandavirus and Uukuvirus, among others [5,1619]. The departments of Córdoba and Cesar in the Colombian Caribbean area are particularly significant due to their geographical conditions, diverse reservoirs (such as rodents and birds), vectors (such as ticks and mosquitoes) and predominantly tropical climatic characteristics (high temperatures and humidity), which facilitate the spread of vectors like ticks and the diseases they transmit [20].
Given the increasing threat of tick-borne viruses to public health, it is imperative to conduct eco-epidemiological surveillance using NGS technologies [21].
Our study, which aimed to identify LITV in the metatranscriptome of hard ticks in the Colombian Caribbean, is a timely and important step in addressing the growing threat of tick-borne diseases. The results of the collected tick species were presented in a recently published article [19].
Methods
Tick capture, taxonomic identification and RNA extraction
Between 2022 and 2023, field trips were conducted in different locations in the departments of Córdoba and Cesar (Fig. 1). Seven hundred and sixty-six ticks were collected and processed. Ticks were collected directly from wild animals (snakes and iguanas) and domestic animals such as cattle, horses, sheep and dogs. Ticks were transported in liquid nitrogen to the laboratory and kept at −80 °C. Specimens were classified using taxonomic keys [22] and grouped by taxonomic genus, sex and stage, with a maximum of 12 individuals. Tick pools were macerated in 600 µl PBS, and 200 µl of the supernatant was transferred to a 22-µm filter. RNA was extracted from the filtrate using the GeneJet RNA Purification Kit from Thermo Scientific™. For sequencing, 13 tick pools were made, by species, genus, stage and geographic location.
Geographic location of tick capture and municipalities sampled with positive results for LITV in the departments of Córdoba and Cesar.
Library preparation and sequencing
The concentration and integrity of the RNA in the pools were measured using the kit IQ assay RNA and BR RNA with Qubit™ (Thermo Fisher Scientific™). Thirteen pools were processed using the MGIEasy RNA Library Prep PE-FLC 150 library preparation kit. The RNA was fragmented into products of ~250 nt. Then reverse transcription of the RNA, second-strand synthesis, end repair and adapter ligation were performed. PCR amplified the latter, and the library was quantified using fluorometry in Qubit™. Fragmentation was assessed using the Agilent Technologies™ Bioanalyzer. ssDNA circularization and DNA nanobead generation for high-throughput sequencing were performed using the MGI-G50 equipment, which generates a throughput of 560 million reads in 32 h (Shenzhen, China).
Bioinformatics and phylogenetic analyses
Initially, the quality of the reads was verified using fastp [23]. The genome sequences of the different tick species analysed (Rhipicephalus sanguineus GenBank accession GCA_013339695.2, Rhipicephalus microplus GenBank accession GCF_013339725.1, Dermacentor nitens GenBank accession GCF_013339745.2 and Amblyomma dissimile GenBank accession GCA_023969395.1) were discarded through reference mapping using Bowtie2 v.2.5.0 [24]. De novo assembly was then performed to obtain contigs with MEGAHIT v.1.2.9 [25], and then the metatranscriptomes were submitted to accelerated blastx in diamond software using the ‘non-redundant’ database of National Center for Biotechnology Information (NCBI), the diamond output were meganized for taxonomic binning in MEGAN6 using a MEGAN mapping file (2022), following the recommendations of the diamond +megan for fast taxonomic analysis [26]. The metatranscriptome contigs matched with viruses were extracted and compared with the current ‘non-redundant’ database of proteins using the online tool blastx 2.14.1 [27]. The sequences identified as viral agents were annotated and translated using Prokka through the Galaxy online platform [2829]. The deduced amino acid sequences of the viral transcripts identified in previous steps were aligned against the amino acid sequences of viral agents retrieved from GenBank [30] with the MAAFT server [31]. Maximum likelihood phylogenetic reconstructions were performed in IQ-TREE v2.2.2.6 with 1,000 ultrafast bootstrap, the best-fitting amino acid substitution model according to ModelFinder [32], phylogenetic trees were visualized in iTOL v.5 [33], and edited in Inkscape v.1.1 [34].
Results
Only 9 of the 13 sequenced groups showed complete and partial fragments of the LITV genome. In the metatranscriptome of R. microplus, D. nitens and A. dissimile, complete and partial fragments of the L segment of LITV collected in the departments of Córdoba and Cesar were detected.
Only in the R. microplus species with code 12G from Cesar was the S segment of LITV found. No fragments of the M segment were detected. In the R. sanguineus species collected in Córdoba, LITV was not detected. Tick species with codes 11G, 29G and 117G were not included in the alignment because the amino acid sequences of the L segment were very short, with a size of 53 aa (Table 1).
Phylogenetic analysis of the L segment showed that LITV from this study is phylogenetically close to LITV (95% ultrafast bootstrap) from Trinidad and Tobago, Brazil, Colombia and the French Antilles. The aforementioned group of sequences LITV American sequences is related to LITV from the Hainan Province in southern China (Fig 2 and S1, available in the online Supplementary Material). In contrast, phylogenetic analysis of the S segment showed that the sequence reported in the study grouped with LITV detected in R. microplus from China and LITV in Rhipicephalus sp. from Thailand (Fig. 2).
Phylogenetic constructs with amino acids from the L and S segments of LITV. (a) A Phylogenetic tree of the L segment encoding RdRp was constructed with 35 sequences (30 were downloaded from GenBank, and 5 were on their own). (b) A Phylogenetic tree of the S segment encoding the nucleocapsid has been built with 30 sequences (29 downloaded from GenBank and 1 own). These two trees were rooted with Brown dog tick mivirus (QDW81054.1) and Manawa virus (AFN73044). The sequences generated in this study are highlighted in red. The trees were constructed using the LG substitution model for the RdRp L and the nucleocapsid S segments.
Analysis of the two segments in blastp showed that the five amino acid sequences corresponding to the L segment have a percentage of identity between 99.86 and 99.32% with the LITV sequence reported in France, Trinidad and Tobago, and Brazil [13536]. Regarding the S segment, the amino acid sequence showed a percentage of identity of 99.32%, compared to the LITV sequence reported in Brazil [1].
Discussion
LITV was identified from R. microplus and D. nitens collected from cattle and A. dissimile collected from iguanas in the Colombian Caribbean. The present work is the fifth report of LITV in Colombia and the first report of this virus in A. dissimile.
Currently, viruses of the Uukuvirus genus are not considered important for public health, although some studies have detected antibodies against some Uukuviruses in humans [1037]. LITV belongs to the Phenuiviridae family, where the viral genus Bandavirus is found, which, in turn, has species of viruses that affect human health, such as B. dabieense and B. heartlandense transmitted by ticks [1112]. It is known that the difference between Bandavirus and LITV is based on the fact that the M segment is absent [337]. In the present study, fragments of the M segment were not detected. These results are supported by several studies conducted in Colombia [5,1618]. LITV was first identified in 2015 in R. microplus from China; the results demonstrated the diversity of viruses in ticks [3]. In 2018 in Brazil, almost complete sequences of LITV from the L and S segments were detected in R. microplus, and these results highlight that LITV has a high distribution frequency. In that study, LITV could not be cultured in vertebrate cell lines [1]. Later in 2019, in Trinidad and Tobago, the virome of R. microplus, R. sanguineus and Amblyomma ovale was analysed; only in R. microplus was LITV found [35].
In contrast, our study found LITV in D. nitens and A. dissimile, demonstrating the vast diversity of tick species that host LITV. In 2020, in the French Antilles, the two segments of LITV were detected in R. microplus from cattle. Phylogenetic analyses showed that the variant is related to the one found in China, suggesting a possible specificity with this tick species [36]. However, there are reports of LITV in D. nitens [17] and those found in our study in A. dissimile. In 2022 in China, LITV was detected in * R. microplus* and R. sanguineus collected from cattle and dogs; in that study, they suggested that LITV could present a low host restriction [38]. In the present study, it was also confirmed in R. microplus, D. nitens and A. dissimile; however, we could not detect LITV in R. sanguineus.
Regarding LITV studies in Colombia, the first report of LITV was in 2020 on R. microplus and D. nitens, collected in cattle and horses, respectively, in the department of Córdoba [17]. This finding differed from the first LITV detected in China in R. microplus [3]. The present study shows the wide range of tick species that carry this virus. In 2020, in the department of Antioquia, partial segments L and S were detected in R. microplus of cattle [16]. In 2021, LITV was detected in R. microplus of cattle and sheep in the middle Magdalena area. Orozco et al. [18] suggest this virus has a wide geographic distribution in R. microplus populations. However, in the present work, LITV was also found in D. nitens and A. dissimile. In addition, LITV was found in the Antioquia department of Amblyomma patinoi and Amblyomma cajennense. The results confirm this virus’s wide geographic distribution in several tick species [5]. It is essential to highlight that LITV has only been found in ticks, unlike other viruses, such as Jingmen, which are found in ticks [19] and mosquitoes [39].
Our results are important because LITV belongs to the Uukuvirus genus and is classified in the Phenuiviridae family [610]. Within this family, there are prominent representatives that affect human and animal health [1112]. In addition, these findings reveal the circulation of LITV in a wide variety of tick species that can parasitize animals and humans.
In conclusion, this study identified LITV in the species D. nitens and R. microplus and is the first report of LITV in A. dissimile. Although LITV has not been considered important in public health, epidemiological surveillance studies in domestic and wild animals parasitized by the tick species involved are necessary. Additionally, cell culture and animal model studies must be implemented to determine the pathogenicity of LITV circulating in the region.
Supplementary material
10.1099/acmi.0.000941.v3Uncited Fig. S1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Souza WM de Fumagalli MJ Torres Carrasco A de O Romeiro MF Modha S et al Viral diversity of Rhipicephalus microplus parasitizing cattle in southern Brazil Sci Rep 2018811010.1038/s 41598-018-34630-130397237 PMC 6218518 · doi ↗ · pubmed ↗
- 2Brackney DE Armstrong PM Transmission and evolution of tick-borne viruses Curr Opin Virol 201621677410.1016/j.coviro.2016.08.00527569396 · doi ↗ · pubmed ↗
- 3Li C-X Shi M Tian J-H Lin X-D Kang Y-J et al Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses Elife 2015412610.7554/e Life.05378 PMC 438474425633976 · doi ↗ · pubmed ↗
- 4Yu X-J Liang M-F Zhang S-Y Liu Y Li J-D et al Fever with thrombocytopenia associated with a novel bunyavirus in China N Engl J Med 20113641523153210.1056/NEJ Moa 101009521410387 PMC 3113718 · doi ↗ · pubmed ↗
- 5Molina-Hoyos K Montoya-Ruíz C Aguilar PV Pérez-Doria A Díaz FJ et al Virome analyses of Amblyomma cajennense and Rhipicephalus microplus ticks collected in Colombia Acta Trop 202425310715810.1016/j.actatropica.2024.10715838402921 PMC 11781606 · doi ↗ · pubmed ↗
- 6Abudurexiti A Adkins S Alioto D Alkhovsky SV Avšič-Županc T et al Taxonomy of the order Bunyavirales: update 2019 Arch Virol 20191641949196510.1007/s 00705-019-04253-631065850 PMC 6641860 · doi ↗ · pubmed ↗
- 7Spiegel M Plegge T Pöhlmann S The role of phlebovirus glycoproteins in viral entry, assembly and release Viruses 2016820210.3390/v 807020227455305 PMC 4974537 · doi ↗ · pubmed ↗
- 8Qin X-C Shi M Tian J-H Lin X-D Gao D-Y et al A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors Proc Natl Acad Sci U S A 20141116744674910.1073/pnas.132419411124753611 PMC 4020047 · doi ↗ · pubmed ↗